Article Text

Abstract
Inflammatory pseudotumour is a generic term applied to a variety of neoplastic and non-neoplastic entities that share a common histological appearance, namely a cytologically bland spindle cell proliferation with a prominent, usually chronic inflammatory infiltrate. Over the last two decades, inflammatory myofibroblastic tumour (IMT) has emerged from within the broad category of inflammatory pseudotumour, with distinctive clinical, pathological and molecular features. IMT shows a predilection for the visceral soft tissues of children and adolescents and has a tendency for local recurrence, but only a small risk of distant metastasis. Characteristic histological patterns include the fasciitis-like, compact spindle cell and hypocellular fibrous patterns, which are often seen in combination within the same tumour. Chromosomal translocations leading to activation of the ALK tyrosine kinase can be detected in approximately 50% of IMTs, particularly those arising in young patients. This review will examine the clinical, pathological, and molecular genetic features of IMT and discuss an approach to diagnosis and differential diagnosis.
Statistics from Altmetric.com
The term “inflammatory pseudotumour” has been used to describe a wide range of reactive and neoplastic lesions, including inflammatory myofibroblastic tumour (IMT), pseudosarcomatous myofibroblastic proliferations of the genitourinary (GU) tract, infectious and reparative processes, and inflammatory pseudotumours of lymph node, spleen and orbit. Over the last two decades, IMT has emerged as a distinct entity with characteristic clinical, pathological and molecular features. However, confusion remains regarding the distinction of these tumours from other lesions in the “inflammatory pseudotumour” family, as well as from non-neoplastic fibrosclerosing processes and malignant neoplasms with a prominent inflammatory infiltrate. This review will examine the clinicopathological and molecular features of IMT and discuss its differential diagnosis, with emphasis on other entities included under the umbrella of inflammatory pseudotumour.
HISTORY
Inflammatory pseudotumour was first described in the lung, where it was considered a reparative postinflammatory condition rather than a neoplastic process.1 2 Histologically similar lesions were subsequently reported at extrapulmonary sites, and distinctive clinical features, including a predilection for children and young adults and associated systemic symptoms in a minority of patients, were recognised.3–8 These tumours were widely considered benign and likely non-neoplastic until the early 1990s when Meis and Enzinger published a series of 38 cases, primarily intra-abdominal and retroperitoneal tumours in children and adolescents, which they termed “inflammatory fibrosarcoma” based on follow-up data showing a significant rate of aggressive behaviour.9 Among the 27 patients with clinical follow-up in this series, 10 (37%) had local recurrences, 3 (11%) developed metastases, and 5 (19%) died from disease.9 In 1995, Coffin et al reported 84 cases of extrapulmonary IMT, which overlapped clinically and histologically with “inflammatory fibrosarcoma,” although the recurrence rate was somewhat lower (25%) and no metastases occurred.10 The identification of recurrent clonal rearrangements involving chromosome 2p over the next several years provided additional support that IMT was a distinct neoplasm; “inflammatory fibrosarcoma” is now considered indistinguishable from, and within the morphological spectrum of, IMT.
CLINICAL FEATURES
IMTs have a predilection for children and adolescents, although they may arise as late as the eighth decade of life.9 11 12 The most common anatomical locations are the abdominopelvic region, lung, and retroperitoneum,10 but virtually any site may be involved, including the somatic soft tissues, bone, larynx, uterus and central nervous system.13–17 Accurate data regarding the incidence and anatomical distribution of IMT are difficult to obtain due to the use of the terms “inflammatory pseudotumour” and “IMT” interchangeably in the literature; however, one recent review of 275 “inflammatory pseudotumours” in children reported that approximately one-third of cases were pulmonary and two-thirds were extrapulmonary.18
Patients generally present with a mass or non-specific symptoms, including vague abdominal pain or gastrointestinal complaints for intra-abdominal lesions, and cough, chest pain, or, less often, haemoptysis for pulmonary tumours.10 19 20 A constitutional syndrome consisting of fever, weight loss and malaise is seen in 15–30% of patients, and laboratory evaluation may reveal microcytic anaemia, a raised erythrocyte sedimentation rate, thrombocytosis, and/or polyclonal hypergammaglobulinaemia.9 10 In some cases, the mass may be found only after an extensive workup for fever of unknown origin or growth failure.5 8 21 The systemic manifestations resolve following surgical excision, and tumour recurrence may be heralded by a return of clinical and laboratory abnormalities.10 19 21 A similar clinical presentation may be seen in the plasma cell variant of Castleman disease, in which overproduction of interleukin 6 (IL-6) is thought to be the underlying cause of the systemic complaints.22 Two patients with IMT were reported to have raised serum levels of IL-6 that returned to normal postoperatively as their systemic symptoms resolved,23 24 and several authors have demonstrated production of IL-6 mRNA and protein by tumour cells, supporting a possible analogous mechanism in IMT.23–25
PATHOLOGICAL FEATURES
Grossly, IMTs may be firm, fleshy, or gelatinous, with a white or tan cut surface. Calcification, haemorrhage and necrosis are identified in a minority of cases.9 10 Tumours range from 1 cm to >20 cm in greatest dimension, with a mean size of 6 cm.9 10 A subset of IMTs, particularly those in the abdomen or retroperitoneum, present as multiple discrete masses in the same anatomical region.7 9 10
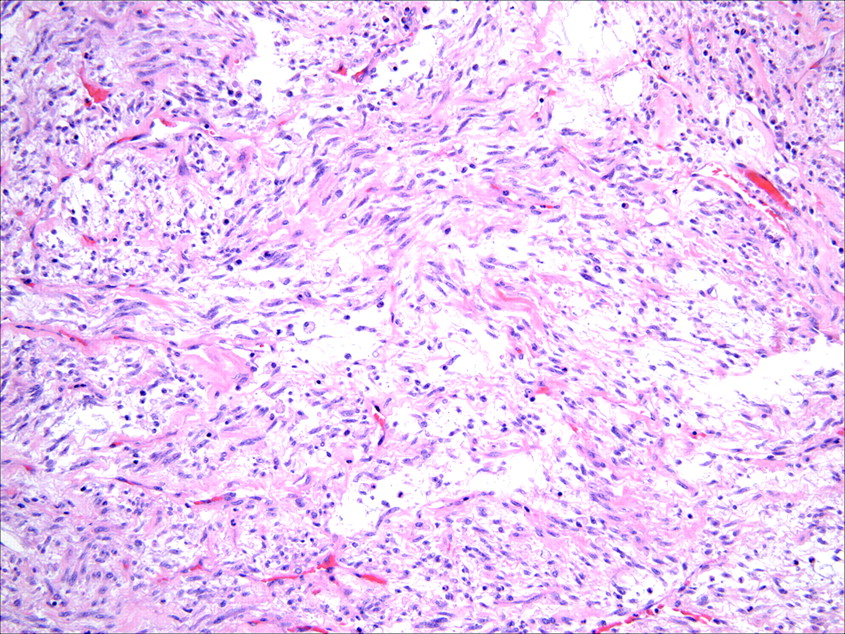
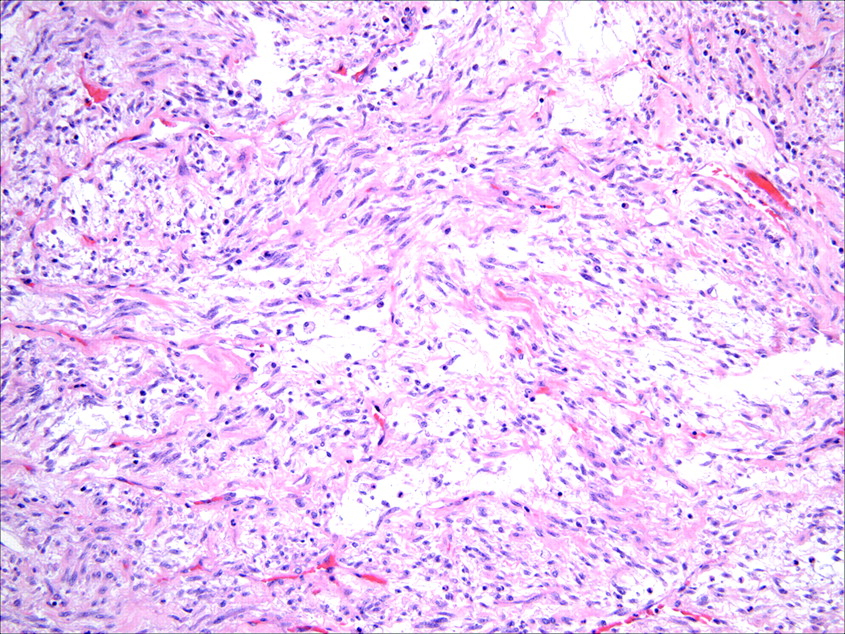
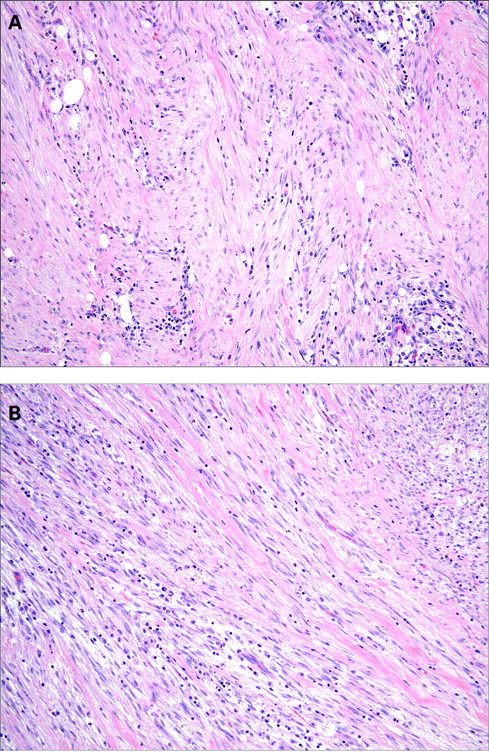
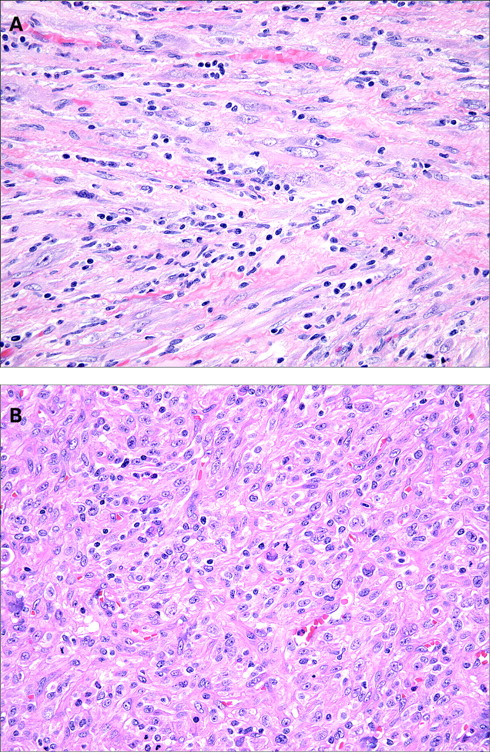
Histologically, IMTs are characterised by a variably cellular spindle cell proliferation in a myxoid to collagenous stroma with a prominent inflammatory infiltrate composed primarily of plasma cells and lymphocytes, with occasional admixed eosinophils and neutrophils. Coffin et al described three basic histological patterns, which are often seen in combination within the same tumour: a myxoid/vascular pattern, a compact spindle cell pattern, and a hypocellular fibrous (fibromatosis-like) pattern.10 The myxoid/vascular pattern has a fasciitis-like appearance, with loosely arranged plump spindle cells in an oedematous or myxoid stroma and a prominent vasculature (fig 1). The inflammatory infiltrate in these areas often contains more neutrophils and eosinophils and fewer plasma cells than in the other two patterns. The compact spindle cell pattern is characterised by a cellular proliferation of spindle cells with a fascicular or storiform architecture in a collagenous stroma (fig 2). These foci typically show numerous plasma cells and lymphocytes intimately admixed with the spindle cells, but discrete lymphoid follicles and aggregates of plasma cells are also common. The fibromatosis-like pattern is relatively hypocellular, with elongated rather than plump spindle cells in a densely collagenous background containing scattered lymphocytes, plasma cells and eosinophils (fig 3). Focal dystrophic calcification and even metaplastic ossification can be seen in hyalinised areas.10 26 Foamy histiocytes are prominent in a minority of IMTs.15 19


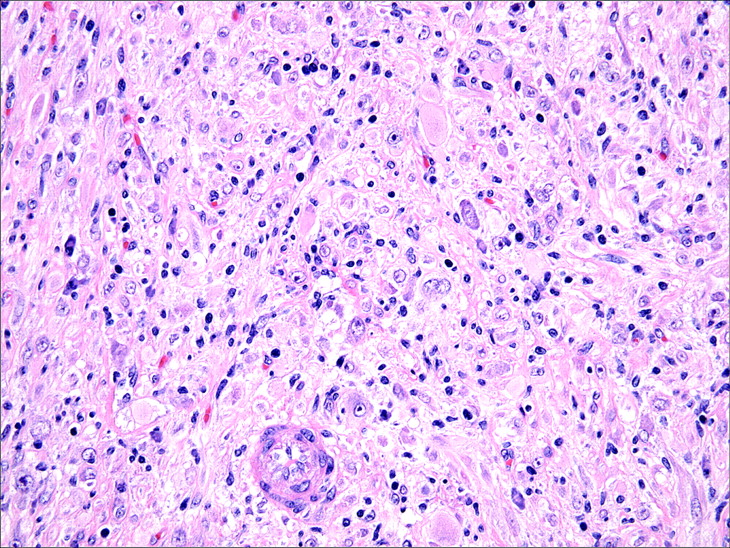
The spindle cells of IMT are typically uniform and predominantly myofibroblastic in appearance, with palely eosinophilic cytoplasm, plump ovoid to tapering vesicular nuclei and one or two small nucleoli (fig 4). Mild nuclear pleomorphism may be seen, but hyperchromasia is absent. Approximately one half of cases contain scattered “ganglion-like” cells (fig 5): larger polygonal cells with abundant amphophilic to eosinophilic cytoplasm, large vesicular nuclei and prominent nucleoli, similar to those seen in proliferative fasciitis.9 27 Mitotic activity is generally low (0–2 mitoses per 10 HPF), and atypical mitoses are rare.10 15 27 28 Necrosis and vascular invasion have been reported in typical IMTs but are very infrequent.10 29 30 Rarely, IMTs may undergo histological evolution to a morphologically higher grade lesion (fig 6) with increased cellularity, marked nuclear atypia, frequent mitoses, atypical mitotic figures, and/or necrosis.10 20 28 31 The cytological features of the morphologically higher grade tumours are variable, including hypercellular spindle cell, epithelioid/histiocytoid, or round cell morphology.10 28 31


By ultrastructural analysis, IMTs are composed predominantly of myofibroblasts with a smaller fibroblastic component10 19 32; the ganglion-like cells show features of fibroblasts.33 As expected given their myofibroblastic differentiation, IMTs are positive for smooth muscle actin in 80–90% of cases and express desmin and calponin in 60–70%, although reactivity for these markers is often focal.10 15 30 33 Approximately one-third of tumours show focal keratin reactivity,10 34 which is not unexpected given that myofibroblasts, similar to smooth muscle cells, may be keratin positive.35 36 The plasma cell infiltrate is polyclonal.15 19
Due to the prominent inflammatory infiltrate and associated systemic symptoms in a minority of patients with IMT, a viral aetiology has been proposed, but the evidence in this regard is unconvincing. A small subset of so-called inflammatory pseudotumours of lymph node, liver and spleen contain detectable Epstein–Barr virus (EBV)37–39; however, it is likely that most if not all of these cases do not represent true IMTs (see below). The presence of EBV in classic IMTs of other anatomical sites is rare.10 33 40–42 One group identified human herpesvirus-8 (HHV-8) DNA in both pulmonary and extrapulmonary IMT, but expression of HHV-8-associated antigens was not investigated,25 43 and other authors have not found evidence for HHV-8 infection in IMT.40 41
MOLECULAR FEATURES
Rearrangements involving the ALK (anaplastic lymphoma kinase) locus on chromosome 2p23 have been documented in both pulmonary and extrapulmonary IMTs, providing further support for the neoplastic nature of these lesions and their distinction from other “inflammatory pseudotumours”.44 45 The ALK gene encodes a receptor tyrosine kinase that is only expressed in neural tissue under normal conditions.46 Clonal abnormalities of ALK were first described in anaplastic large cell lymphoma (ALCL), in which 50–60% of cases contain a translocation of the ALK gene leading to constitutive tyrosine kinase activation.47 Approximately 50% of IMTs also show ALK rearrangements by FISH,44 and several ALK fusion partners have been identified, including TPM3 at 1p23, TPM4 at 19p13, ATIC at 2q35, CLTC at 17q23, CARS at 11p15, RANBP2 at 2q13 and SEC31L1 at 4q21.48–54 The most common fusion protein in ALCL (NPM-ALK) has not been identified in IMT, but the TPM3-ALK, ATIC-ALK and CLTC-ALK fusion proteins have been reported in both neoplasms, and thus represent rare examples of translocation-derived chimeric tyrosine kinases driving both mesenchymal and lymphoid neoplasms.55–59
By immunohistochemistry, approximately 50% of IMTs are positive for ALK, with reactivity in several large series ranging from 36% to 71% (fig 7). Similar to ALCL, ALK expression in IMT is more common in younger patients, but is not restricted to this population.11 44 50 60 ALK expression in IMT reliably predicts the presence of an ALK gene rearrangement (which can be detected by FISH or RT-PCR),11 42 44 and the pattern of ALK immunostaining may correlate with the specific gene fusion,11 although this has yet to be validated in large series of ALK-positive cases. Localisation of ALK within the cell appears to be determined by its fusion partner, leading to diffuse cytoplasmic staining for ALK with the TPM3, TPM4, CARS, ATIC and SEC31L1 fusion partners, all of which are cytoplasmic proteins11 51 52 54; nuclear membrane staining with the RANBP2 fusion partner, a nuclear pore protein11 53; and granular cytoplasmic staining with the CLTC fusion partner, a main structural protein of coated vesicles.11 49 Although current data are too limited to draw conclusions regarding a possible relationship between gene fusion type and prognosis, both reported cases with the RANBP2-ALK fusion showed round cell transformation, raising the possibility that this subset may be clinically more aggressive.11 53

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Most other myofibroblastic and fibroblastic tumours, including desmoid fibromatosis, nodular fasciitis, calcifying fibrous tumour, myofibromatosis, and infantile fibrosarcoma, are negative for ALK.11 61 62 ALK expression has been reported in a subset of malignant peripheral nerve sheath tumours, rhabdomyosarcomas, Ewing sarcomas, leiomyosarcomas, extraskeletal myxoid chondrosarcomas, and benign and malignant adipocytic neoplasms,11 61 63 but with the exception of leiomyosarcoma, these tumours generally do not enter the histological differential diagnosis of IMT. The molecular mechanisms responsible for ALK expression in non-IMT mesenchymal tumours are uncertain; most cases lack ALK rearrangements by RT-PCR or FISH and express the full-length ALK transcript rather than the truncated or chimeric form seen in IMTs.61
PROGNOSIS
IMTs are classified as tumours of intermediate biological potential by the most recent World Health Organization classification, due to a tendency for local recurrence and a small risk of distant metastasis.34 The recurrence rate varies by anatomical site, from <2% for tumours confined to the lung18 64 to 25% for extrapulmonary lesions.10 Recurrences are particularly common among multinodular intra-abdominal tumours and those in delicate anatomical locations such as the larynx or trachea, likely reflecting the difficulty of complete surgical resection. In contrast, several recent studies have reported that recurrence is very infrequent following complete excision of a solitary lesion.64–67
Distant metastasis of IMT is rare, occurring in <5% of cases.34 Of the 21 metastasising IMTs in the literature, the primary tumours affected patients over a broad age range (17 months to 79 years) and arose in a variety of anatomical sites, including 7 in the lung, 6 in the abdomen/mesentery, 3 in the retroperitoneum, and 1 each in the mediastinum, liver, iliac bone, neck, and forearm.9 12 28 31 32 51 68–75 The most common sites of metastasis are lung and brain, followed by liver and bone. Metastatic disease is usually identified at presentation or within a year of diagnosis, but occasional patients develop metastases as many as 9 years following excision.74
Several studies have attempted to identify histological predictors of aggressive behaviour in IMT without much success. Tumour size, cellularity, mitotic activity and the presence of necrosis do not appear to correlate with outcome.27 28 30 One study suggested that the presence of nuclear atypia and ganglion-like cells might indicate more aggressive behaviour, but these features were also seen in over half of the clinically benign IMTs, limiting their prognostic utility.27 Furthermore, in a recent study of atypical and/or clinically aggressive IMTs, half of the metastasising cases showed no atypical features.28 In contrast to ALCL, in which ALK-positive tumours pursue a less aggressive clinical course, there is no clear-cut relationship between ALK expression and prognosis in IMT.28 30 44 75 Recent studies suggest that ALK-positive tumours have a very low risk of metastasis (if any), but ALK reactivity does not appear to correlate with recurrence.28 44 75 The role of p53 reactivity in predicting behaviour of IMTs is uncertain, as the frequency of p53 immunoexpression has ranged from <10% to 80% in various studies, and some investigators have shown a correlation between p53 positivity and aggressive behaviour, while others have not.27 28 30 DNA aneuploidy as determined by flow cytometry may be associated with an increased risk of recurrence,27 32 76 but this technique is not widely available.
DIAGNOSTIC APPROACH AND DIFFERENTIAL DIAGNOSIS
Table 1 lists several key points to consider when confronted with a spindle cell lesion containing a prominent inflammatory infiltrate. These points should be considered general (not invariant) guidelines.
In our experience, IMTs are more commonly overdiagnosed than underdiagnosed. Attention to the clinical context will often prevent this error—in particular, one should be wary of making the diagnosis of IMT in a middle-aged or elderly patient. The anatomical site also provides clues, since these tumours have a strong predilection for visceral organs and the deep soft tissues of the abdomen, pelvis, and retroperitoneum. IMT should be considered a diagnosis of exclusion in the skin and superficial somatic soft tissues, lymph node, spleen or bladder, where close histological mimics are more common (see discussion below). The histological differential diagnosis of IMT depends in part on the dominant pattern—myxoid/vascular, compact spindle cell or fibromatosis-like. In general, immunohistochemistry does not play a major role in confirming the diagnosis, due to the variable expression and lack of specificity of myofibroblastic markers. ALK positivity is helpful if present, but its absence does not exclude the diagnosis of IMT, particularly in adults. We will first consider the specific differential diagnosis of IMT based on histological pattern, and then discuss criteria for the diagnosis at several specific anatomical sites, focusing on those in which the distinction between “inflammatory pseudotumour” and IMT is poorly defined.
The compact spindle cell pattern of IMT is particularly difficult to distinguish from a variety of histological mimics. Occasional spindle cell sarcomas, spindle cell melanomas, and sarcomatoid carcinomas contain a marked inflammatory infiltrate, and may show only mild cytological atypia.77–79 However, plasma cells are generally not a prominent component of the inflammatory infiltrate in these other tumour types, and most examples show at least focally prominent nuclear hyperchromasia, atypical mitoses, necrosis or vascular invasion, all of which are very unusual in IMT. Dedifferentiated liposarcoma can be particularly challenging, especially on a biopsy specimen, as morphologically low grade lesions are often remarkably bland,80–82 and immunohistochemistry for MDM2, a sensitive marker of dedifferentiated liposarcoma,83 is not helpful in this distinction, given that a significant proportion of IMTs show nuclear MDM2 expression.28 30 However, dedifferentiated liposarcoma usually presents in an older age group than IMT, and adequate sampling of the mass and adjacent soft tissue will usually reveal a higher grade component and/or areas of well-differentiated liposarcoma. Due to their predominance among mesenchymal tumours of the GI tract and mesentery, gastrointestinal stromal tumours (GISTs) may be considered in the differential diagnosis of cellular IMTs in the abdomen. However, the palely eosinophilic, syncytial cytoplasm and cytological uniformity of GISTs contrast with the plump myofibroblasts, scattered ganglion-like cells and collagenous background seen in IMT. Furthermore, although an inflammatory infiltrate may be seen in GISTs, it is generally patchy, and plasma cells are infrequent. Immunohistochemistry is helpful in this differential diagnosis, as IMTs are consistently negative for c-kit.11 84 Other entities to consider in the differential diagnosis of cellular IMTs are dendritic cell neoplasms, which characteristically show an evenly distributed chronic inflammatory infiltrate admixed with the spindle cell component.85 Follicular dendritic cell (FDC) and interdigitating dendritic cell (IDC) sarcomas are readily distinguished from IMT by immunohistochemistry, as the former express CD21 and/or CD35, and the latter are uniformly positive for S100 protein. Fibroblastic reticulum cell (FBRC) tumours are exceptionally rare with only few reports in the literature, most of which have arisen in lymph nodes, where IMT is very uncommon.85 86 FBRC tumours may be histologically indistinguishable from IMT and have a similar immunoprofile, including variable expression of SMA, desmin and keratin. Indeed, Rosai et al have recently proposed that IMTs show fibroblastic reticulum cell, rather than myofibroblastic, differentiation,87 but this hypothesis remains speculative. Finally, inflammatory leiomyosarcoma, although a rare histological variant, deserves mention, as it shares several features with IMT, including a predilection for young adults, a mixed storiform and fascicular spindle cell architecture, and a prominent inflammatory infiltrate, often with numerous foamy histiocytes.88 However, focal areas with typical histological features of leiomyosarcoma—namely, fascicles of spindle cells with cigar-shaped nuclei and brightly eosinophilic cytoplasm—can usually be identified.
Fibromatosis-like or hypocellular fibrous IMTs may show histological overlap with desmoid fibromatosis or calcifying fibrous tumour, depending on the degree of stromal hyalinisation. Desmoid fibromatosis, particularly if located in the mesentery, not infrequently shows focally fasciitis-like features, enhancing the histological resemblance to IMT. In classic areas, however, the spindle cells of fibromatosis are arranged in characteristic long fascicles, in contrast to the short fascicular or storiform patterns of IMT, and although a lymphocytic infiltrate may be present in desmoid tumours, plasma cells are infrequent. Furthermore, these lesions generally show aberrant nuclear positivity for β-catenin.89 Calcifying fibrous tumour is a rare benign neoplasm with a predilection for young patients and a wide anatomical distribution. Some authors have proposed that this lesion represents a late stage of IMT,90 while others have found no convincing link between the two entities.62 91 Histologically, calcifying fibrous tumours are uniformly hypocellular, in contrast to the variable cellularity of IMTs, and they usually contain scattered psammomatous or dystrophic calcifications.
The principal entity in the differential diagnosis of IMTs dominated by the myxoid/vascular pattern is nodular fasciitis. Clinical information is often helpful in this differential, as nodular fasciitis usually appears rapidly over a few weeks to months, is rarely larger than a few centimetres in size, and typically arises in subcutaneous tissue or skeletal muscle, where IMT is very uncommon. Histologically, IMTs contain a much more prominent inflammatory infiltrate than nodular fasciitis and usually show collagenous storiform or fascicular areas, which are not seen in the latter. Occasionally, reactive processes dominated by granulation tissue may mimic IMTs with the myxoid/vascular pattern. In the absence of a supportive clinical history, histological clues to a reactive/post-traumatic process include an organised vascular pattern and fat necrosis in the adjacent soft tissue. Fasciitis-like IMTs arising in the GU tract should be distinguished from pseudosarcomatous myofibroblastic proliferations (see below).
Inflammatory pseudotumour versus IMT
As discussed above, IMT was formerly buried within the broad category of non-neoplastic fibroinflammatory and neoplastic lesions referred to as inflammatory pseudotumour. The use of these terms synonymously in the literature has led to confusion regarding the true incidence and behaviour of, and the diagnostic criteria for, IMT at a variety of sites, particularly the lung, liver, spleen, lymph node, and bladder.
Pulmonary inflammatory pseudotumour (also often referred to as plasma cell granuloma) encompasses a variety of entities, including IMT and post-infectious/reparative processes, of which the latter are likely more common.2 20 92 93 Twenty to 30% of patients with pulmonary inflammatory pseudotumour report a history of lower respiratory tract infection, while others have a history of pulmonary infarcts or prior radiation therapy.19 20 92 94 Organising pneumonia is frequently identified within or at the periphery of these lesions,20 92 and many cases show features not seen in IMT, such as granulomatous inflammation, abscess formation, and numerous lymphoid follicles with germinal centres.92 A recent study reported increased IgG4-positive plasma cells and obliterative phlebitis in a series of plasma cell-rich pulmonary inflammatory pseudotumours, suggesting that some cases may have an autoimmune aetiology and form part of the spectrum of the recently recognised “systemic IgG4-related sclerosing disease”.95–97 The broader age distribution and lower recurrence rate for pulmonary IMT as compared to extrapulmonary tumours18 64 98 may be due in part to the inclusion of non-neoplastic processes in prior studies. That said, the lung remains a common site for true IMT, and, conversely, IMT is one of the most common primary pulmonary neoplasms in children.94 99
Similar to those in the lung, hepatic “inflammatory pseudotumours” represent a diverse group of lesions with infectious, autoimmune and neoplastic aetiologies, the latter including inflammatory pseudotumour-like FDC sarcoma and IMT. A bacterial or fungal cause has been identified in several cases of hepatic inflammatory pseudotumour, and in some reports, the masses resolved following antibiotic therapy.100–102 Prior reports of EBV in hepatic inflammatory pseudotumours are now believed primarily to represent inflammatory pseudotumour-like FDC sarcomas.103–105 This distinctive variant of FDC sarcoma characteristically arises in the liver and spleen of female patients, frequently presents with systemic symptoms, and shows a consistent association with EBV.105 Histologically, inflammatory pseudotumour-like FDC sarcomas are composed of spindle cells with vesicular nuclei and palely eosinophilic cytoplasm and show a marked lymphoplasmacytic infiltrate, thus closely mimicking IMT. Immunohistochemistry is often required to distinguish between these two neoplasms: the spindle cells of inflammatory pseudotumour-like FDC sarcoma express at least one of the FDC markers (CD21, CD23, and CD35), are negative for ALK, and are consistently positive for EBER (EBV-encoded mRNA).105 Finally, recent studies suggest that hepatic inflammatory pseudotumours dominated by lymphoplasmacytic inflammation with a minimal myofibroblastic component may be a manifestation of the systemic IgG4-related sclerosing disease (see above).106 107 Similar to the pancreatic lesions associated with this disorder (“autoimmune” or lymphoplasmacytic sclerosing pancreatitis), the “lymphoplasmacytic type” of hepatic inflammatory pseudotumour consistently shows increased IgG4-positive plasma cells, obliterative phlebitis, and periductal inflammation with concentric fibrosis, whereas these findings are less common or absent in the “fibrohistiocytic type” of hepatic inflammatory pseudotumour.107
Inflammatory pseudotumour of lymph node and spleen is a non-neoplastic entity distinct from IMT. Most affected patients are adults, and those with lymph node lesions frequently present with systemic symptoms and/or laboratory abnormalities similar to those seen in a minority of patients with IMT.108–111 Inflammatory pseudotumour of lymph node has a characteristic distribution, preferentially involving the connective tissue framework of the node (the capsule, trabeculae, and hilum) without forming a discrete mass, although in later stages, the lesion may efface the nodal architecture.110 Vascular changes, including perivascular fibrosis and vasculitis, are often seen within medium-sized vessels of the hilum or capsule, and extension into perinodal soft tissue is common.109–111 EBV has been detected in small lymphocytes in several cases of nodal inflammatory pseudotumour, but the spindle cells are EBV negative.37 109 In contrast to nodal lesions, splenic inflammatory pseudotumours are often asymptomatic, form a discrete mass, and usually lack associated vascular changes.39 108 109 Furthermore, splenic lesions are rarely associated with nodal lesions and vice versa, suggesting that the two entities may be biologically unrelated, despite their histological similarities.108 Some investigators have detected EBV in the spindle cells of a subset of splenic inflammatory pseudotumours,37–39 whereas others reported no evidence of EBV infection.109 The spindle cells in some of the EBV-positive cases also expressed FDC markers,37 39 and thus represent EBV-associated inflammatory pseudotumour-like FDC tumours, as discussed above. Inflammatory pseudotumours of lymph node and spleen are consistently negative for ALK.11 39 109
The family of idiopathic fibrosclerosing lesions, including sclerosing mesenteritis, idiopathic retroperitoneal fibrosis, sclerosing mediastinitis, and orbital inflammatory pseudotumour, may also be considered within the umbrella of inflammatory pseudotumour. These lesions may occur synchronously or metachronously in the same patient.112 113 Recently, an association with elevated serum IgG4 levels and other autoimmune disorders, particularly autoimmune pancreatitis, has been described in some patients with fibrosclerosing diseases, suggesting that a subset of these cases may be manifestations of the systemic IgG4-related sclerosing disease mentioned above in the discussion of pulmonary and hepatic inflammatory pseudotumours.96 97 114 Grossly, the mass lesions formed by these processes are ill-defined and often encase adjacent structures, in contrast to the relatively circumscribed margins of IMT. Microscopically, the spindle cell component of fibrosclerosing lesions is usually significantly less cellular than that seen in even the most hypocellular IMTs, and the storiform, fascicular and myxoid/vascular patterns of IMT are absent. In addition, the inflammatory infiltrate is often patchy and perivascular, in contrast to the diffuse infiltrate seen in IMTs, and lymphocytic infiltration of blood vessel walls may be present in up to 50% of cases.113 115
Finally, perhaps the most confusing topic in the recent literature on IMT is the distinction between pseudosarcomatous myofibroblastic proliferations (PMPs) and IMTs in the GU tract. Pseudosarcomatous spindle cell lesions in the GU tract have been described under a variety of names, including pseudosarcomatous fibromyxoid tumour, inflammatory pseudotumour, pseudosarcomatous myofibroblastic tumour/proliferation, pseudomalignant spindle cell proliferation, and postoperative spindle cell nodule for those arising following instrumentation.116–123 There are no histological differences between those that occur following trauma and those that arise spontaneously, the latter being more common.124 125 PMP usually occurs in adults, is not associated with systemic symptoms, and may recur locally in 10–20% of cases but does not metastasise.124 125 Although some authors do not distinguish between IMT and PMP in the GU tract,126 127 we and others42 124 125 128 believe that they are distinct entities that are separable on morphological grounds, and, in our experience, most fasciitis-like myofibroblastic lesions in the GU tract are PMPs. Histologically, PMPs are composed of a haphazard to loose fascicular arrangement of spindle cells, many of which have elongated bipolar cytoplasmic processes that are often brightly eosinophilic, mimicking rhabdomyoblasts.120 121 124 The stroma is typically oedematous to myxoid with prominent vascularity and a variably dense acute and/or chronic inflammatory infiltrate.124 125 PMP may show focally more cellular fascicular areas, particularly in the deeper portion of the lesion away from the mucosal surface, but the storiform and hypocellular fibrous patterns of IMT are rarely seen. In addition, the inflammatory infiltrate of PMP is generally less dense than that seen in IMT and lacks a prominent plasma cell component.124 125 As both IMT and PMP are myofibroblastic in nature, there is considerable immunohistochemical overlap between the two lesions. PMP expresses SMA in approximately 70% of cases, desmin in 35–60%, keratin in 42–94%, and may also be positive for ALK, although the molecular basis for this is unclear.124 125 One study documented ALK positivity in 10 of 21 PMPs, but found no evidence of ALK gene rearrangement in the 6 ALK-positive cases examined by FISH.124 Another group similarly found ALK expression in nearly 50% of PMPs (12 of 26) but also identified ALK gene rearrangements in 4 of the 6 ALK-positive cases examined, raising the possibility that these lesions may be neoplastic.125 Alternatively, this study (and others) may have included a heterogeneous group of lesions, both PMPs and “true” IMTs. However, there are certainly rare examples of bona fide IMTs that arise in the region of the bladder. Although the aetiology of PMP (reactive or neoplastic) and its possible relationship to IMT remain controversial, we believe there is sufficient data with respect to differences in clinical behaviour to warrant their separation.
In summary, IMT is a distinctive myofibroblastic neoplasm that has a predilection for the lung, abdomen, and pelvis of children and young adults, shows a tendency for local recurrence, and shows a characteristic cytogenetic abnormality in approximately half of the cases. IMTs should be distinguished from the variety of neoplastic and reactive lesions included under the umbrella term “inflammatory pseudotumour”. To avoid ambiguity, the latter designation is best avoided, with the exception of inflammatory pseudotumour of lymph node and spleen and orbital inflammatory pseudotumour, which are distinct clinicopathological entities. Finally, one should keep in mind that IMT is a diagnosis of exclusion in middle-aged or older adults, and in skin and somatic soft tissue, and the presence of nuclear hyperchromasia, atypical mitoses, or more than mild nuclear atypia argues strongly against the diagnosis.
Take-home messages
The unqualified designation “inflammatory pseudotumour” is best avoided.
Inflammatory myofibroblastic tumour is a distinctive neoplasm of intermediate biological potential with a predilection for the abdominopelvic region and lung of children and young adults.
Histological features in inflammatory myofibroblastic tumours do not correlate well with clinical behaviour.
Chromosomal translocations leading to activation of the ALK tyrosine kinase (and overexpression of the ALK protein) can be detected in approximately 50% of IMTs, but are uncommon in older patients.
Inflammatory myofibroblastic tumour is a diagnosis of exclusion in middle-aged or older adults, and in somatic soft tissue, and the presence of more than mild nuclear atypia argues against the diagnosis.
REFERENCES
Footnotes
Competing interests: None declared.