
Abstract
The National Comprehensive Cancer Network guidelines for the treatment of stage IV metastatic melanoma state that clinical trials should be the preferred treatment option for these patients; therefore, clinical trials for these patients are the “standard of care.” Our greater understanding of the molecular biology behind the development of melanoma has guided much of the translational research in this disease and has led to the development of novel targeted therapies. Specifically, advancement in our knowledge of alterations in signal transduction pathways in melanoma has led to the rapid development of a number of pharmacologic agents that inhibit these pathways. This review will discuss changes in signal transduction pathways involved in melanoma, specifically, activated mitogen-activated protein kinase, activated PI3 kinase, and inactivated p53/Rb pathways. This article will also review new targeted therapy in the context of the molecular biology of melanoma.
INTRODUCTION
Treatment of metastatic melanoma beyond the regional lymph node basin with standard therapies has not been shown to improve the median overall survival of 9 months. The only therapies in current use that are approved by the Food and Drug Administration are dacarbazine (DTIC) and high-dose interleukin-2. DTIC's low response rates on the order of 10% to 15% and virtually no complete responses make this treatment far from ideal.1,2 The second approved therapy, high-dose interleukin-2, also has extremely low response rates and significant toxicity despite its approximately 16% overall response rate and 6% to 8% complete response rate.3 Given these numbers, it is clear that novel treatments are desperately needed, and clinical trials exploring these new agents are the new standard of care for the treatment of metastatic melanoma.
Melanoma originates from melanocytes, and the biology of these cells may give clues to the pathophysiology of melanoma. Cutaneous melanocytes originate from neural crest progenitors that migrate to the basal layer of the epidermis during embryonic development. Epidermal keratinocytes regulate melanocyte homeostasis via secretion of factors that regulate survival, differentiation, and proliferation. Ultraviolet radiation stimulates the release of these factors from keratinocytes, thus ultimately causing the release of melanin from melanocytes.4 During the evolution from benign melanocyte to malignant melanoma, this tight-knit regulation from keratinocytes is lost.5 The lost communication can lead to a benign nevus; however, with continued lack of regulation a malignant melanoma can develop.
The 4 main clinical types of melanoma (superficial spreading, lentigo maligna, nodular, and acral lentiginous) may all have a unique and different pathophysiologic and molecular basis. One study used genome-wide alteration in DNA copy number combined with individual somatic mutations to distinguish between types of melanoma, with 70% accuracy.6 As our understanding of the molecular biology of melanoma and its subtypes evolves, a more targeted approach to the treatment of this disease develops. The Table lists a number of novel agents classified by the primary target of each agent.
Targeted Therapies in Melanoma
MOLECULAR PATHWAYS
Cell Cycle
The cell cycle of growth and division is a carefully coordinated process that is distinctly deregulated in melanoma via deregulation of cyclins, cycle-dependent kinases (CDKs), and cell-cycle inhibitors.7 Cyclins bind the CDKs, which lead to the phosphorylation of downstream proteins that ultimately cause cell-cycle progression.
The most common aberration to cell-cycle control in melanoma affects the CDKN2A genetic locus. Figure 1 demonstrates this gene, its products, and how these products interact with the cell cycle. This locus controls the RB1 and p53 pathways.7 The CDKN2A locus is located on chromosome 9, which is also the chromosome that has been linked to familial melanoma.8,9 It is estimated that CDKN2A mutations increase susceptibility to melanoma as well as other cancers such as pancreatic adenocarcinoma. CDKN2A contributes to 10% to 40% of familial melanoma cases.10,11
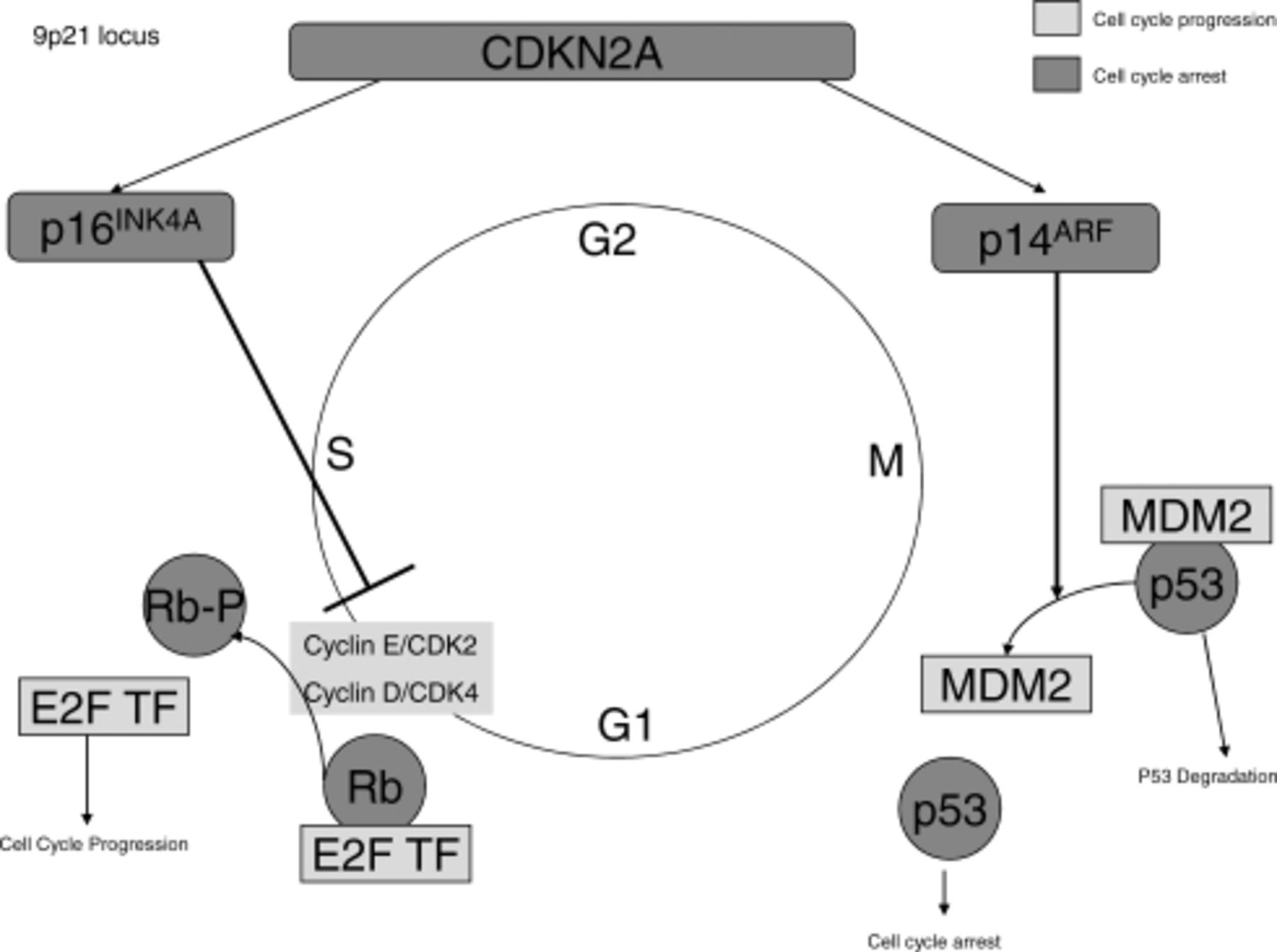
Cell cycle is controlled by the Rb pathway and p53 pathway. The CDK2NA gene encodes for p16 and p14. p16 inhibits the cyclins from phosphorylating Rb. Phosphorylated Rb cannot bind and inhibit E2F transcription factor. p14 controls MDM2. MDM2 cannot bind and negatively regulate the p53 when p14 is bound to it.
This gene codes for 2 tumor suppressor proteins, p16INK4A and p14ARF. These proteins negatively regulate the cell cycle via the RB1 pathway and the p53 pathway, respectively (Figure 1). The RB1 pathway controls the transition from G to S phase of the cell cycle via E2F transcription factors that lead to the transcription of S-phase genes. Whereas p53 regulates senescence and cell-cycle arrest, the protein p14ARF binds MDM2. MDM2 protein negatively regulates p53 via targeting it for degradation. Loss of function of either p16INK4A or p14ARF leads to increased growth and proliferation.7
Approximately 50% of melanomas have CDKN2A locus mutations, leading to loss of function of p16INK4A. Both deletion of the gene and silencing methylation can lead to lack of gene expression of p16INK4A, with methylation occurring in 20% to 75% of melanomas.12,13 Methylation of the promoter region of the gene prevents transcription of the gene, leading to lack of expression of the gene product. This leads to loss of negative regulation of the cell cycle, thus causing increase in proliferation.
Even though loss of function in p53 via p14ARF leads to the same results as loss of RB1, it occurs in a vastly different pathway. MDM2 negatively regulates p53, and MDM2 function is regulated by p14ARF. Direct mutations in p53 are infrequent in melanoma, occurring in fewer than 10% of humans. Inactivation of this pathway occurs more via CDKN2A and loss of function of its product p14ARF.7,14
Therapeutic targets manipulating the cell cycle have been a challenge to develop. Demethylation agents such as 5-azacytidine and 5-aza-2′-deoxycytidine (decitabine) are being explored both alone and in combination with high-dose interleukin-2. When decitabine was combined with interleukin-2, objective responses occurred in 31% of melanoma patients.15 Restoring loss of tumor suppressor proteins like p16 and p14 has been difficult; therefore, most efforts are directed at targeting CDK. Flavopiridol is an inhibitor of CDK 1, 2, 4, and 6. UCN-01 inhibits both CDK2 and protein kinase C. Both drugs have been in early clinical trials for patients with melanoma but have not been successful because of toxicities as well as limited clinical efficacy as single agents. No objective clinical responses were seen in metastatic melanoma patients treated with flavopiridol in a phase II study.16 MDM2 inhibitors, such as nutlin, are also in phase I trials. More specific CDK inhibitors are under development.
Mitogen-Activated Protein Kinase Pathway
The mitogen-activated protein kinase (MAPK) pathway links growth factor receptors to intracellular signaling pathways via activation of downstream kinases (Figure 2). MAPK signaling is initiated via binding of receptor tyrosine kinases. These lead to the activation of RAS, a small G protein with 3 isoforms (HRAS, KRAS, NRAS) located on the inner surface of the plasma membrane of a cell. A number of receptor tyrosine kinases interact with RAS, including epidermal growth factor receptor, c-KIT, platelet-derived growth factor receptor, vascular endothelial growth factor receptor, fibroblast growth factor receptor, and FLT-3.17

The mitogen-activated protein kinase (MAPK) pathway and PI3K pathways. Drugs that inhibit specific components of the pathways are noted.
RAS, when activated, can complex with another protein kinase, RAF, that also has 3 isoforms ARAF, BRAF, and CRAF. This activated complex leads to the phosphorylation of MAPK (also known as ERK) via activation of MEK. MAPK, when phosphorylated, can directly enter the nucleus and effect translation of genes. This ultimately leads to the control of cellular proliferation (Figure 2).
Mutations in the pathway are abundant in melanoma. Activating mutations of BRAF are seen in 50% to 60% of melanomas and are also frequently present in benign nevi. However, most nevi do not transform into malignant melanoma.6,18 This implies that BRAF mutation may be necessary but not sufficient to induce malignant transformation. The most common mutation seen is the V600E mutation in which a glutamine is substituted for a valine.18 This leads to constituently stimulated MAPK pathway, thus leading to increased survival and growth.
RAS mutations are also present in a number of melanomas. Mutations in NRAS are most common and involve approximately 15% to 30% of cases. This also leads to a constitutively active MAPK pathway. NRAS and BRAF mutations appear to be mutually exclusive, suggesting redundancy in the pathway.18–20
Targeted drugs manipulating this pathway are an intense area of clinical research. Sorafenib is a multikinase inhibitor that targets BRAF as well as a number of other kinases, including CRAF, vascular endothelial growth factor receptor-2, vascular endothelial growth factor receptor-3, and platelet-derived growth factor receptor.16 Downstream targets of BRAF are clearly decreased with the addition of sorafenib in vitro, with studies showing decreased MAPK phosphorylation.21 However, the in vivo studies thus far have been disappointing. Little or no antimelanoma activity has been shown with sorafenib alone in phase I/II clinical trials.22 Sorafenib in combination with chemotherapy demonstrated high response rates (89 of 135 patients with complete response, partial response, or stable disease) in clinical trials, but no definitive evidence of improved overall survival or progression-free survival in the second-line setting for stage IV melanoma patients.23-25 Eastern Cooperative Oncology Group (ECOG) 2603 evaluated the role of sorafenib in combination with carboplatin and paclitaxel in the front-line setting and did not meet the primary endpoint of improved overall survival in 800 patients with unresectable stage III or IV metastatic melanoma.26 However, a phase II study did find a significant improvement in progression-free survival (but not overall survival) when sorafenib was combined with DTIC chemotherapy in chemo-naïve patients.27
More specific BRAF inhibitors are under development. RAF-265 is a drug that inhibits both RAF kinase and vascular endothelial growth factor receptors. In melanoma cell lines harboring either NRAS or BRAF mutations, RAF-265 potently inhibits the MAPK pathway. Also in xenograft models, this drug showed tumor regression whereas sorafenib only led to tumor stabilization.27
PLX-4032 is another BRAF inhibitor with a high affinity for mutated BRAF. Recently presented phase I data on PLX-4032 showed that 9 of 16 patients demonstrated tumor regression in stage IV BRAF-mutant melanoma, with a median duration of response of 8.5 months.28 These data are extremely promising and there is much excitement over these results. The phase II trial of this drug recently completed enrollment, and the final results are eagerly awaited. A phase III trial comparing PLX-4032 to DTIC is planned.
MEK inhibitors, such as AZD6244 and PD0325901, are also in early-phase trials. PD0325901 is not being moved forward secondary to unexpected toxicity and poor pharmacologic properties.29 AZD6244 monotherapy was compared in a phase II study with temozolomide, and there was no significant difference in progression-free survival between those arms. However, there were patients documented to have lasting remissions to AZD6244, mainly those patients with documented BRAF mutations.30 AZD6244 is currently being evaluated in combination with chemotherapy. Many trials also have correlative studies evaluating tumor samples for target inhibition.
PI3K/AKT Pathway
Although the MAPK pathway leads to proliferation, the PI3 kinase (PI3K) pathway is prosurvival via antagonizing the intrinsic pathway of apoptosis (Figure 2).31 PI3K is activated by growth factor receptors, which thus lead to the activation of AKT via PDK1. AKT can then prolong survival by directly activating transcription factors that lead to the transcription of prosurvival genes.31 This process is antagonized by PTEN when PTEN leads to apoptosis rather than survival. PI3K pathway also regulates cell proliferation via control of entry into the cell cycle at the G1-checkpoint.32 FOXO is another antagonist of the AKT pathway and FOXO proteins can be directly inactivated by AKT.33 mTOR is another downstream effecter of AKT that ultimately leads to enhanced cell growth.
Although no distinct mutations in PI3K and AKT have been found in melanoma, the PI3K pathway is constitutively active in melanoma.34 This pathway may be activated in melanoma via paracrine or autocrine activation of growth factor receptors. This has been demonstrated with insulin-like growth factor-I, which is shown to increase growth of early-stage melanoma cells via the PI3K pathway.35,36 The other way that the PI3K pathway is altered is through loss of negative regulation. PTEN is the main negative regulator and is lost in approximately 30% of melanoma cell lines and 10% of human melanoma tumors.37,38
A number of inhibitors of this pathway are in the early stages of development, all targeting this pathway from different angles. Perfosine inhibits AKT phosphorylation and translocation of AKT to the cell membrane. However, this drug produced no objective responses as a single agent in a phase II trial in metastatic melanoma.39, Another route of inhibition of this pathway comes from an inhibitor of PDK-1. PDK-1 is critical in AKT activation and is inhibited by UCN-01.18 Phase II trials in melanoma have been completed with this drug, but results are pending. Various mTOR inhibitors are also being evaluated in patients with melanoma. A phase II study of temsirolimus had only one partial response lasting just 2 months.41 Another mTOR inhibitor, RAD-001, demonstrated 7 patients with stable disease and is now in the second stage of evaluation.42
c-KIT
Many other signaling pathways are altered in melanoma. The c-KIT gene encodes a receptor tyrosine kinase, which can be activated by stem cell factor (also called KIT ligand). Interestingly, c-KIT receptor is necessary for melanocyte survival but c-KIT expression is lost in benign nevi.7 KIT deregulation occurs preferentially in acral and mucosal melanomas as well as melanomas that occur on chronically sun-damaged skin.43
Curtin et al43 demonstrated that mutations or amplification of KIT was seen in 39% of mucosal membranes, 36% of acral skin, and 28% of melanomas on chronically sun-damaged skin. None were found in melanomas without chronic sun damage. Of note, none of these melanomas harbored BRAF mutations. This study found that 11 of the melanomas had KIT mutations that affected the juxtamembrane domain of KIT, thus leading to constitutive activation via dimerization even in the absence of ligand.
Given this information, it is logical that imatinib (Gleevec) should be evaluated in these subtypes of melanoma. Imatinib is a small-molecule tyrosine kinase inhibitor that potently inhibits the kinase activity of BCR-ABL, mutated c-KIT, and platelet-derived growth factor receptor.44 In a small pilot trial, and in a number of anecdotal publications, blockade of c-KIT with the oral inhibitor Gleevec induced rapid and dramatic regression in patients with c-KIT mutated mucosal melanoma. A phase II trial of imatinib in metastatic melanoma patients demonstrated one patient with a partial response lasting approximately 1 year. This patient had acral lentiginous melanoma and had the highest c-KIT expression of the 21 patients in this study.45 Studies are ongoing evaluating the role of imatinib in patients with acral, mucosal, and melanomas on chronically sun-damaged skin. Studies are also evaluating whether wild-type, mutant, or amplified c-KIT predicts response to imatinib. Other inhibitors of c-KIT are also under investigation.
ALTERNATIVE PATHWAYS
Several other pathways are also commonly altered in melanoma. These include alterations in Bcl-2, nuclear factor-kB (NF-kB), as well as dysregulation of the β-catenin pathway.
NF-kB is a transcription factor that is composed of 2 subunits of the REL protein family, a 65 and 50 kDA subunit. It is activated by cytokines such as tumor necrosis factor-alpha and interleukin-1. NF-kB in normal cells is suppressed in the cytoplasm by inhibitory proteins called IkB. These cytokines allow for phosphorylation and degradation of the 26S proteosome.36 This allows NF-kB to enter the nucleus and activate gene expression, thus permitting control over cell survival and regulation of apoptosis. Increased activity of this pathway can occur via upregulation of the p50 and Rel A subunits or via activation of the PI3/AKT or MAPK pathways.46 Loss of E-cadherin expression is linked to upregulation of NF-kB activity as well. This has been directly implicated in melanoma development. Loss of E-cadherin expression and upregulation of N-cadherin is one of the events by which melanocytes escape regulatory control from keratinocytes.5,
Many of the signaling pathways discussed in this article affect NF-kB, thus making this an ideal therapeutic target. Bortezomib (PS-341) targets NF-kB indirectly by inhibition of the 26S proteosome, which leads to increased levels of the IkB inhibitory subunit. In vitro studies have been promising, demonstrating both reduced cell-line growth and synergism with chemotherapy.48 No single-agent activity has been seen with PS-341, but combination studies with temozolomide are ongoing. Early studies with temozolomide and PS-341 showed 4 of 19 patients with some regression of tumor.49
Other targets in melanoma include Bcl-2 and HSP90. Bcl-2 is important in normal melanocyte development with melanoblasts and melanocytes expressing high levels of Bcl-2. Bcl-2−null mice, in fact, have loss of melanocyte numbers.50,51 Drug resistance in melanoma has been at least partially attributed to Bcl-2 by inhibiting apoptosis. Chemotherapy triggers cell death by activating an apoptotic cascade that is initiated by mitochondrial release of cytochrome C. Cytochrome C release is inhibited by Bcl-2, thus inhibiting apoptosis. Studies have demonstrated that transfection of cells with Bcl-2 confers a multidrug resistant phenotype. The converse has also been shown to increase response to chemotherapy.52-54
Oblimersen, an antisense oligonucleotide directed against Bcl-2, thus decreases expression of Bcl-2 by blocking the translation from mRNA. More than 750 patients with metastatic melanoma were randomized to receive dacarbazine or dacarbazine plus oblimersen. Progression-free survival was improved by 1 month with the addition of the study drug, and overall survival, although not statistically significant, was improved by 1.2 months. Of note, in patients with a normal lactate dehydrogenase level, survival was statistically better in the study group, 11.4 months versus 9.7 months.2 The AGENDA trial was thus conducted comparing DTIC with oblimersen versus DTIC alone in metastatic melanoma patients with a normal lactate dehydrogenase. This trial did not show a statistically significant difference between the 2 groups regarding progression-free survival. Although the study reported numerical differences in progression-free survival, overall response, disease control, and durable response that favored the group that received oblimersen, none of these differences were statically significant.55
Another area of investigation is targeting heat shock protein 90 (HSP90). HSP90 is a ubiquitously expressed chaperone that regulates posttranslational folding and stability of proteins. Several oncogenic proteins depend on HSP90, including BCR-ABL, EGFR, RAF, and AKT.56 Thus, inhibiting HSP90 would allow for inhibition of several different signal transduction pathways including the MAPK and PI3K pathways. HSP90 expression is found to be associated with adverse histologic features of melanoma and in metastatic melanoma.57 Prolonged stable disease has been seen in early clinical trials of HSP90 inhibition, and trials with HSP90 inhibitors are ongoing.58
Elesclomol is an experimental agent that induces oxidative stress and is a potent inducer of heat shock proteins. A phase II randomized controlled trial of weekly elesclomol plus paclitaxel versus paclitaxel alone in 53 patients showed a doubling of the median progression-free survival in the treatment arm (112 vs 56 days) and an improvement in median overall survival as well (11.9 vs 7.8 months).59 A phase III clinical trial of weekly paclitaxel plus or minus elesclomol in metastatic melanoma patients was halted in February 2009 based on the recommendation of the Data Monitoring Committee (DMC) to unblind the study after the DMC observed increased deaths on the experimental treatment arm; however, the DMC could not determine whether the observed deaths were related to treatment. The results of the 651 patients enrolled were recently published and demonstrated a median progression-free survival of 3.5 months with elesclomol and paclitaxel versus 1.9 months in the paclitaxel alone arm (HR 0.88), which was not statistically significant.60
CONCLUSIONS
Melanoma biology is an extremely complex process involving multiple mutations as well as activation of multiple oncogenic pathways. Clearly, targeting one pathway is not likely to be clinically effective in treating patients with advanced melanoma. Studies are underway combining different targeted agents, as well as with combinations of chemotherapy and/or biologic therapy and signal transduction inhibitors. Clinical trials with well-conducted correlative studies will contribute to our understanding of the molecular biology of melanoma. Improved understanding of melanoma biology and its mechanisms of resistance to current treatments may potentially lead to the development of newer, more effective agents.
- Academic Division of Ochsner Clinic Foundation
REFERENCES
In this issue








{kind=link}
{kind=link}
Jump to section
Cited By...
- No citing articles found.