
ABSTRACT
Background A fundamental problem in the identification of new molecular targets for therapeutic intervention in diabetic nephropathy has been the lack of an experimental mouse model that faithfully recapitulates human diabetic nephropathy.
Methods Our laboratory, in collaboration with Drs Kakoki and Smithies at the University of North Carolina-Chapel Hill, has developed novel strains of Akita diabetic mice in which the p66 longevity gene has been deleted by homologous recombination. We chose to delete p66 because p66 controls mitochondrial metabolism and cellular responses to oxidative stress, aging, and apoptosis. The redox function of p66 is indispensable for the exponential increase in reactive oxygen species (ROS) associated with diabetes.
Results p66 null Akita mice express a protection phenotype in kidneys that includes marked attenuation of oxidative stress and glomerular/tubular injury and a striking reduction in urine albumin excretion. Furthermore, the p66 null mutation not only confers a survival advantage to podocytes but also prevents foot process effacement and retains the stationary phenotype. Sirtuin 1 (SIRT1) deacetylase and p66 share overlapping biological functions but induce divergent phenotypes, including opposite effects on longevity, ROS metabolism, cell senescence, and apoptosis. Exciting new data from our laboratory show that SIRT1 is upregulated in the kidneys of p66 null Akita mice and decreases acetylation of p53, which destabilizes the p53 protein and prevents the transcription of p53 proapoptosis genes. Conversely, SIRT1 activates the transcription of FOXO3a-dependent stress gene programs that detoxify ROS and promote the survival phenotype.
Conclusion We will focus future research on translating these experimental findings in the mouse to clinical diabetic nephropathy.
INTRODUCTION
The prototypical lesion of diabetic nephropathy is mesangial expansion. However, in advanced stages, glomeruli are virtually void of cells, having been replaced by extracellular matrix. Massive proteinuria and progressive decline in glomerular filtration rate also characterize the late phase of diabetic nephropathy, requiring the initiation of renal replacement therapy. In the United States, the cost for end-stage renal disease (ESRD) is projected to reach $40 billion by 2015. While this cost projection is grim, it does not reflect the true picture of the disease because it does not account for the immense human pain and suffering associated with this complication of diabetes.
The sequential discovery of angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) in the 1980s and 1990s revolutionized the approach to diabetic patients with renal disease.1 Angiotensin II blockade offers the advantage of lowering systemic blood pressure while selectively decreasing glomerular capillary pressure via the relaxation of the efferent arteriole.2 Clinically, these effects are translated into reduced urinary albumin excretion. Although the effectiveness of angiotensin II blockade remains uncontested, ACE inhibitors and ARBs do not stop or prevent the progression to ESRD. Thirty years after the discovery of these agents, diabetes remains the number one diagnosis for new dialysis-dependent patients.
Multiple lines of evidence indicate that genetic predisposition3-6 and oxidative stress7-9 are critical determinants in the development of diabetic nephropathy. Candidate genes have been identified that confer increased risk for diabetic nephropathy, including polymorphisms in ACE and mutations at bradykinin 1 (B1) and bradykinin 2 (B2) receptor loci. These data support a role for the autacoid bradykinin in the renoprotective effect of ACE inhibitors. Bradykinin, via its cognate receptor, promotes the production of nitric oxide that functions as a free radical scavenger, attenuating intracellular oxidative stress.7-9 Accordingly, mutations at the B1/B2 receptor loci or polymorphisms in ACE that result in increased metabolism or breakdown products of bradykinin7-9 may contribute to the oxidative burden in the diabetic kidney.
MICE MODELS
The absence of an experimental animal model that faithfully mimics human diabetic nephropathy has been an obstacle to the identification of novel molecular targets for therapeutic intervention in this disorder. The mouse genome is tractable and accessible to manipulation. For these reasons, genetically engineered mice provide an invaluable resource to dissect the molecular biology of human diseases. Mice and rats, unlike other mammals, have 2 functional insulin genes located on separate chromosomes.10 The Ins1 gene arose from a duplication of the Ins2 gene more than 20 million years ago and has been retained in the genome of mice and rats. The Ins2 gene is orthologous to the human insulin gene. A dominant mutation in the Ins2 gene of Akita mice, caused by an amino acid change, leads to misfolding of the insulin protein and results in hyperglycemia and diabetes. Akita mice have several advantages over inbred mouse strains that require streptozotocin treatment to induce diabetes.4 These advantages include a better defined etiology (endoplasmic reticulum stress and proteotoxicity in pancreatic β cells) and more pronounced and durable hyperglycemia.
Kakoki and Smithies at the University of North Carolina-Chapel Hill have identified a pivotal role for B1 and B2 receptors in the development of diabetic nephropathy in Akita mice. A detailed analysis shows that Akita mice lacking the B2 receptor (Akita B2R-/-) at 6 months of age demonstrate marked enhancement of mesangial expansion and sclerosis, resembling that seen in human diabetic glomerulosclerosis, and significant increases in urine albumin excretion (Figure 1).4 These Akita mice also show greater levels of oxidative stress, mitochondrial DNA damage, and premature expression of senescent-associated phenotypes (alopecia, osteoporosis, thinning skin, fat distribution, and apoptosis). Deletion of both B1 and B2 receptors adds to the severity of the injurious phenotypes. Our search for a model to test whether gene-based interventions can delay or prevent diabetic nephropathy fostered a collaboration between the 2 laboratories.

Light micrographs showing periodic acid-Schiff staining of glomeruli counterstained with hematoxylin from Akita (Ins2+/C96Y/B2R+/+) and Akita (Ins2+/C96Y/B2R-/-) genotypes. B2R, bradykinin 2 receptor.
OUR RESEARCH
In previous work,11,12 we provided evidence that signaling molecules of the insulin-like growth factor 1 (IGF-1) pathway activate gene programs in resident glomerular cells maintained under hyperglycemic conditions that promote the survival phenotype. This work led us to hypothesize that redox-sensitive components of the IGF-1 signaling pathway, such as Src homology 2 domain-containing, transforming protein 1 (p66), may be targets for therapeutic intervention. The p66ShcA protein is one of 3 isoforms encoded by the mammalian ShcA locus. The 3 overlapping Shc proteins—p66ShcA, p52ShcA, and p46ShcA (Figure 2)—all share a C-terminal Src homology 2 (SH2) domain, a central collagen-homologous region, and an N-terminal phosphotyrosine binding domain. p46ShcA and p52ShcA are products of alternative translation initiation sites within the same transcript, whereas p66ShcA is distinguished by a unique N-terminal region generated from alternative splicing.13 p66 also increases reactive oxygen species (ROS) production in the cytosolic compartment via activation of the Rho GTPase Rac1, which amplifies the redox function of NADPH oxidases. Finally, the potent stress response regulator FOXO3a is a downstream target of p66 redox signals that phosphorylate and inactivate FOXO3a via an evolutionary conserved Akt/protein kinase B–dependent pathway. Taken together, p66 controls intracellular ROS metabolism at multiple sites and is a major determinant of cell redox status.

Modular organization of ShcA isoforms. CH, collagen-homologous; PTB, phosphotyrosine binding domain; SH, Src homology.
To test whether inactivating mutations at the p66 locus will rescue kidneys from diabetic nephropathy, we backcrossed SV129/p66-/- mice 6 generations to mice with the C57BL/6J background carrying the Ins2+/C96Y mutation (Akita) and the Ins2+/C96Y/B2R-/- mutations. The p66 null mutation confers a strong protection phenotype in the kidneys of p66 knockout Akita mice and in the kidneys of Akita mice expressing all 3 mutations (Ins2+/C96Y/B2R-/-/p66-/-), as judged by histology (Figure 3) and urine albumin excretion (Figure 4). Histologically, tubular injury and interstitial fibrosis are well documented in advanced diabetic nephropathy. We find that these lesions are markedly attenuated in the kidneys of Akita mice lacking p66 (Figure 5), whereas Akita mice lacking the B2 receptor show the most advanced histological lesions.

Periodic acid-Schiff (PAS) staining of kidney sections. The Ins2 mutation alone increases accumulation of PAS-positive matrix at 12 months of age, which is enhanced by the absence of the bradykinin 2 receptor. The p66 null mutation attenuates renal histological changes at corresponding intervals: nodular PAS-positive matrix (black arrows in Akita and double mutant Akita [DMA]), mesangiolysis (yellow arrows in Akita and DMA), tubular dilation (blue arrow in DMA), and microaneurysm (arrowhead in DMA). The scale bar is 10 μm. The insert shows the histologic analysis of glomerulosclerosis by semiquantitative morphometric analysis. Results are presented as mean ± standard deviation; n=5 in each group; P<0.01 Akita (Ins2+/C96Y) vs p66 knockout (KO) Akita (p66-/-/Ins2+/C96Y); P<0.001 DMA (Ins2+/C96Y/B2R-/-) vs triple mutant Akita (TMA) (p66-/-/Ins2+/C96Y/B2R-/-). WT, wild type.
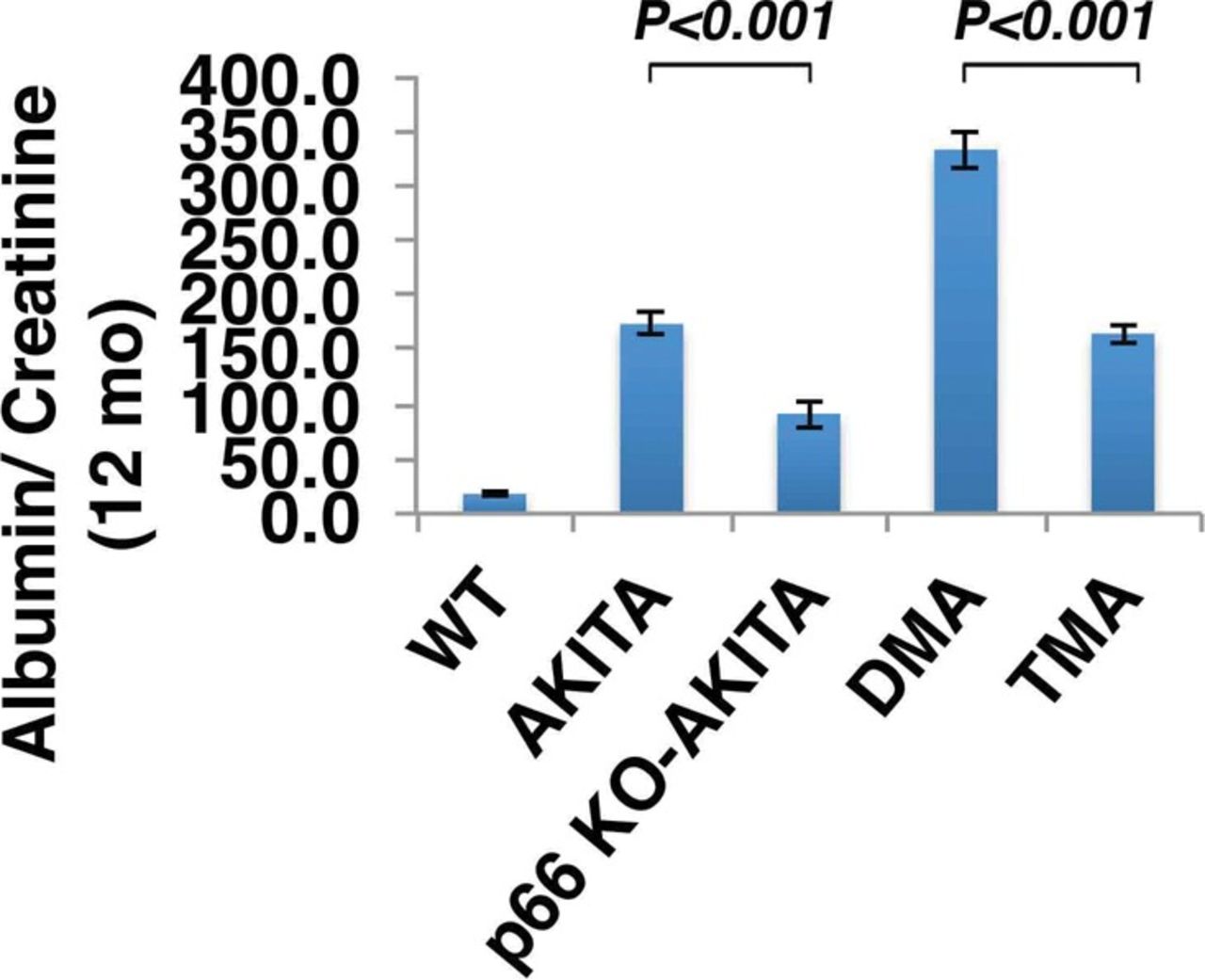
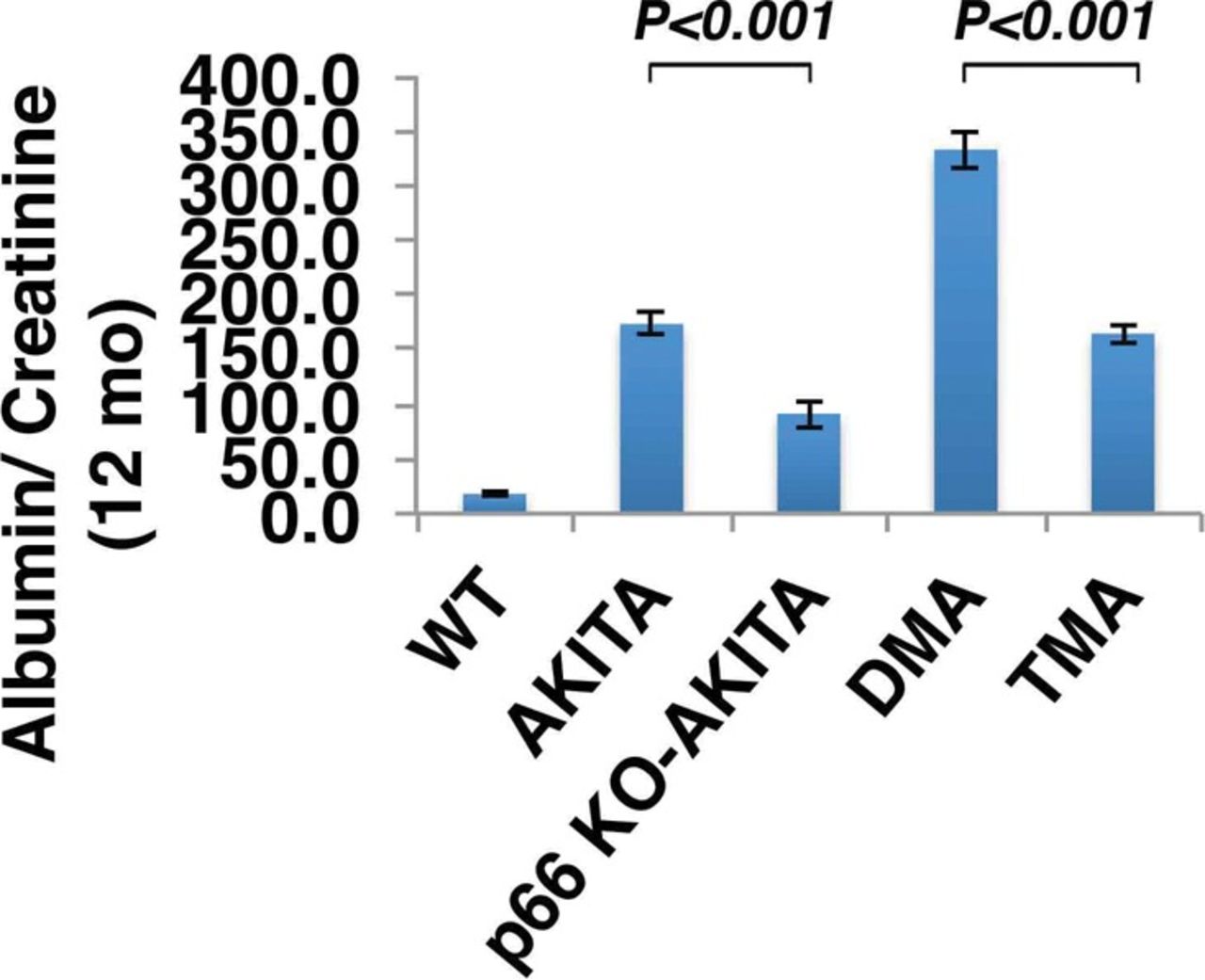
The p66 null mutation attenuates urine albumin excretion. Results are presented as mean ± standard deviation; n=5 in each group. P<0.001 Akita (Ins2+/C96Y) vs p66 knockout (KO) Akita (p66-/-/Ins2+/C96Y); P<0.001 double mutant Akita (DMA) (Ins2+/C96Y/B2R-/-) vs triple mutant Akita (TMA) (p66-/-/Ins2+/C96Y/B2R-/-).
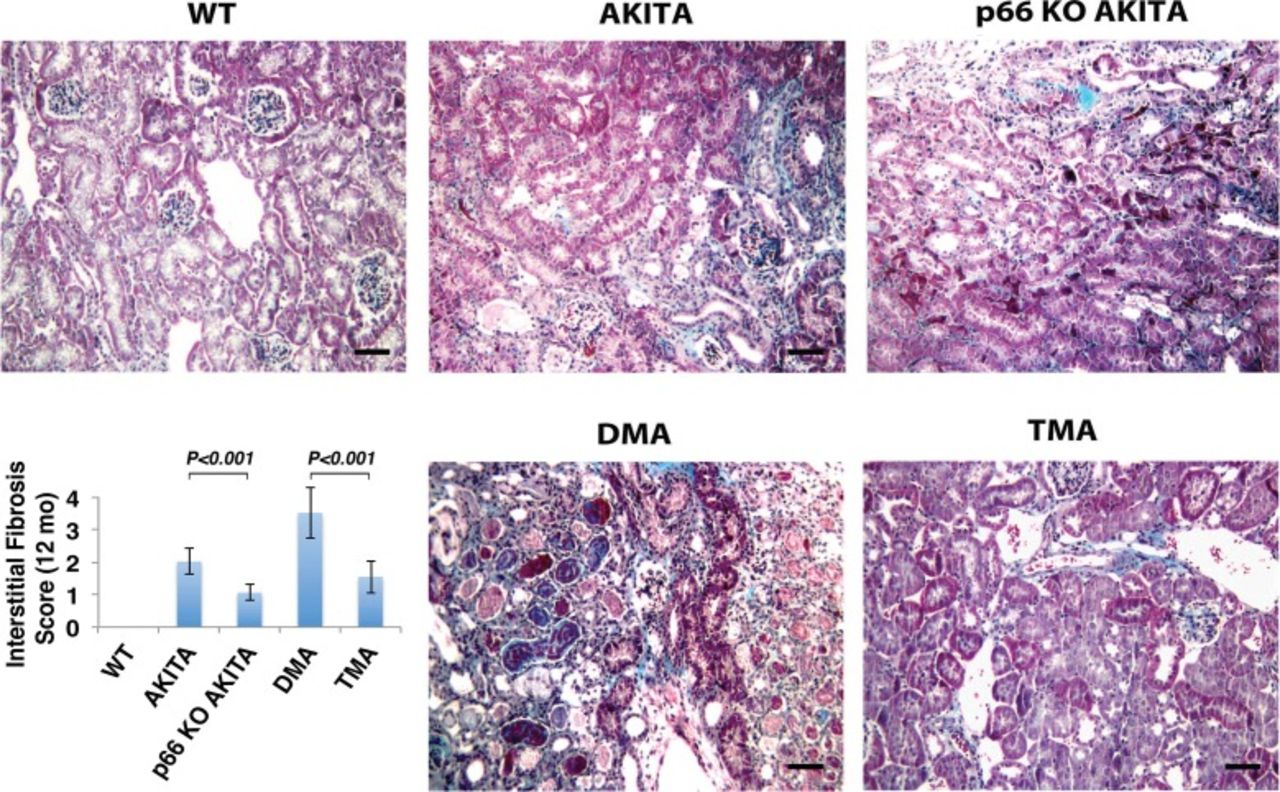
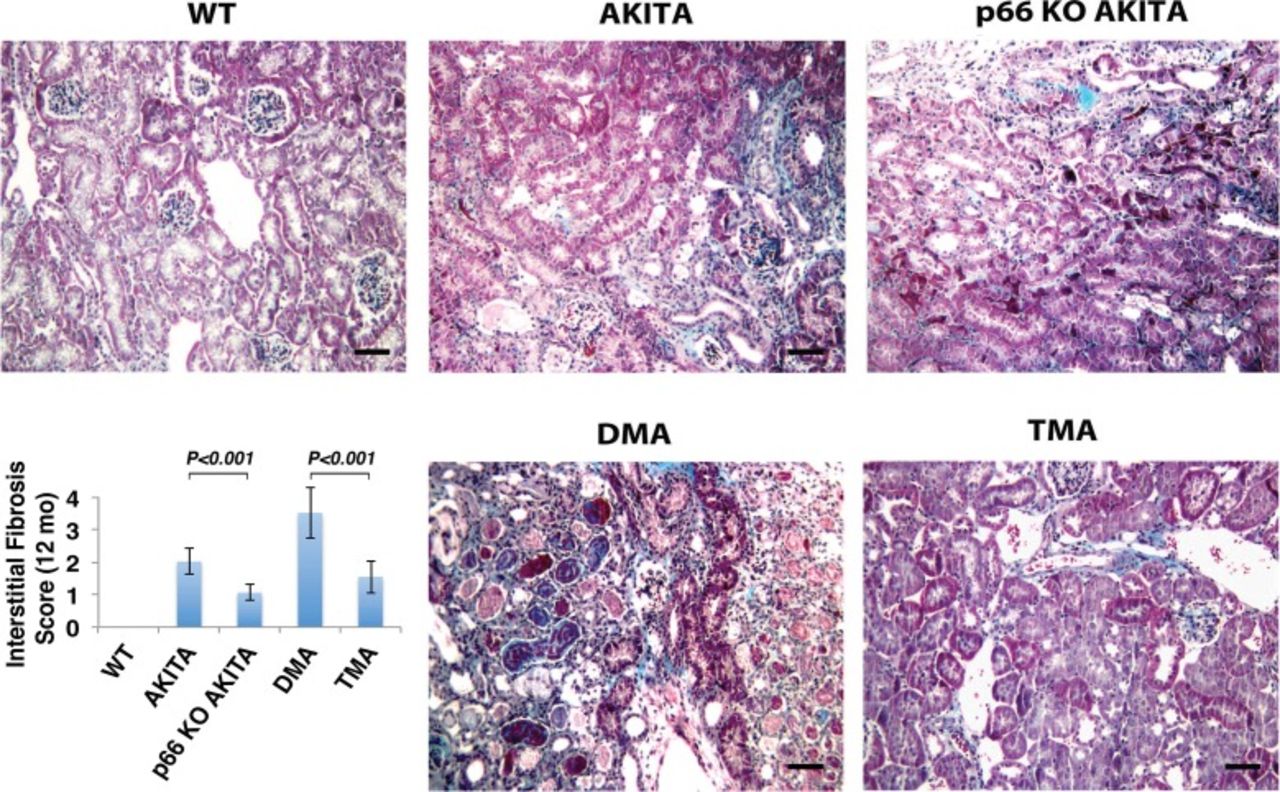
Trichrome staining of the renal cortex. Double mutant Akita (DMA) mice (Ins2+/C96Y/B2R-/-) show increased interstitial fibrosis at 12 months of age, whereas the p66 null mutation attenuates interstitial fibrosis at corresponding intervals. The scale bar is 50 μm. The insert shows the histologic analysis of interstitial fibrosis assessed by semiquantitative evaluation. Results are presented as mean ± standard deviation; n=5 in each group. P<0.001 Akita vs p66 knockout (KO) Akita; P<0.001 DMA vs triple mutant Akita (TMA) (p66-/-/Ins2+/C96Y/B2R-/-). WT, wild type.
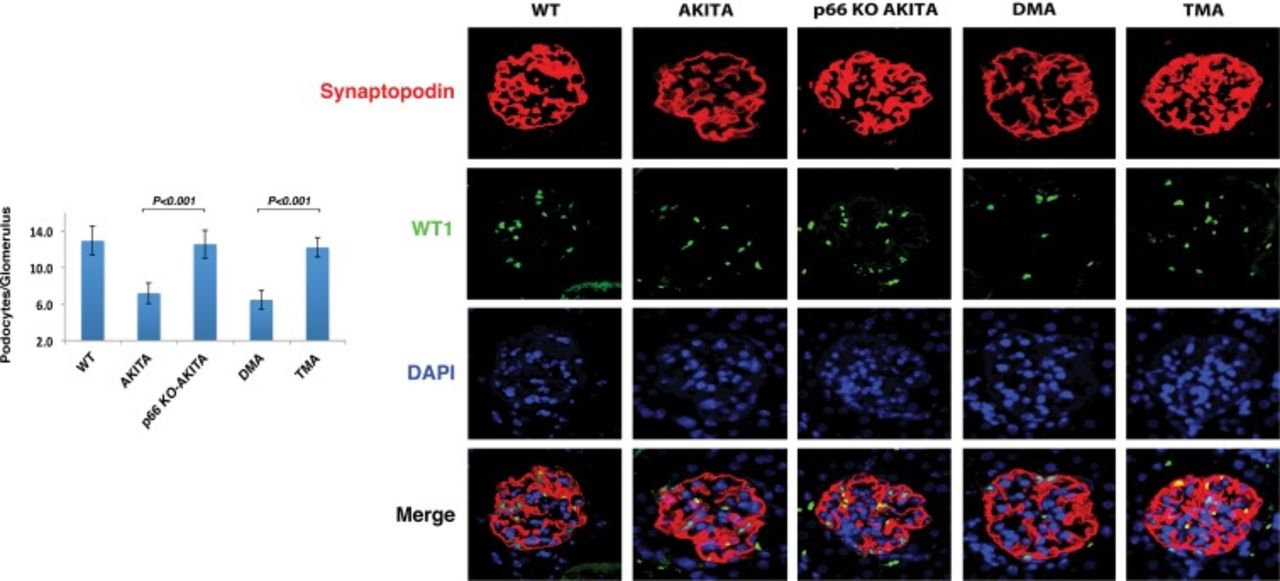
The glomerular visceral epithelial cell, or podocyte, is a key target cell in diabetic nephropathy. Podocytes are the final barrier to the passage of protein into the urinary space. A major limitation in the development of innovative therapy to arrest or prevent diabetic nephropathy is the absence of podocyte regeneration following apoptosis. To date, homozygous mutation at the p66 locus is the only intervention demonstrated to rescue podocytes in the diabetic glomerulus. We found that podocyte number/glomerulus at 12 months of age in the kidneys of p66 knockout Akita mice and mice carrying all 3 mutations was equivalent to age-matched wild-type littermates and was 2-fold greater than that of Akita mice expressing p66 (Figure 6). Importantly, podocyte effacement or loss of foot processes was barely detectable in the kidneys of p66 null Akita mice, perhaps accounting for the striking reduction in urine albumin excretion.

Podocyte number/glomerulus. WT1 (Wilms tumor 1) is expressed in the podocyte nucleus only. Marker-expressing nuclei were counted in the kidneys of Akita mice. Results are presented as mean ± standard deviation; n=5 in each group. Thirty glomeruli from each mouse were analyzed; P<0.001 p66 knockout (KO) Akita (p66-/-/Ins2+/C96Y) vs Akita (Ins2+/C96Y); P<0.001 double mutant Akita (DMA) (Ins2+/C96Y/B2R-/-) vs triple mutant Akita (TMA) (p66-/-/Ins2+/C96Y/B2R-/-). Confocal images of glomeruli show the podocyte-specific marker synaptopodin (red), podocyte nuclear marker WT1 (green), and nuclear counterstain DAPI (blue) in kidney sections at 12 months of age. Merge shows the expression of synaptopodin, WT1, and DAPI.
To elucidate the mechanisms by which the inactivation of p66 confers protection in the diabetic kidney, we turned to our in vitro system of conditionally immortalized differentiated human podocytes (CIDHPs). Podocyte cell lines stably transfected with isoform-specific p66 shRNA (CIDHP/p66 shRNA) were maintained under hyperglycemic conditions and were generated and loaded with redox-sensitive fluoroprobes. Silencing p66 inhibits high glucose (HG)-induced ROS metabolism in mitochondrial and cytosolic compartments while also interrupting the HG apoptosis signal (Figure 7).

(A) Representative immunoblot analysis of ShcA isoforms in conditionally immortalized differentiated human podocytes (CIDHPs) stably transfected with p66 shRNA. (B) CIDHP/p66 shRNA shows inhibition of HG-induced reactive oxygen species (ROS) metabolism. (C) Quantitative analysis of ROS. (D) CIDHP/p66 shRNA shows inhibition of HG-induced apoptosis signal, *P<0.01. EV-CIDHP, empty vector-transduced conditionally immortalized differentiated human podocytes; HG, high glucose (40 mM); Manni, mannitol; NG, normal glucose (5 mM); OD, optical density.
Finally, ongoing work in our laboratory shows an unexpected interaction between the longevity genes sirtuin 1 (SIRT1) deacetylase and FOXO3a in the kidneys of Akita mice lacking p66 (Figure 8). Overexpression of SIRT1 that represses transcription of p66 has been previously reported.14 We found that inactivation mutations at the p66 locus upregulate endogenous SIRT1 and its deacetylase activity. The proapoptosis gene p53 is a substrate of SIRT1 that deacetylates the p53 protein, a modification that destabilizes and inactivates the p53 protein. Conversely, the potent stress response regulator FOXO3a is also a substrate of SIRT1 and results in the transcription of FOXO3a stress gene programs that detoxify ROS and promote DNA repair.

Representative immunoblot analysis of sirtuin 1 (SIRT1) deacetylase and acetylation of lysine-382 of p53 protein. SIRT1 expression is downregulated in Akita diabetic mice but restored in p66 knockout mice. Similarly, the expression of acetyl p53 is upregulated in Akita diabetic mice but returns to normal expression levels in p66 knockout mice. β-actin was used as a loading control. KO, knockout; WT, wild type.
CONCLUSION
Future directions will focus on translating our experimental findings in the mouse to clinical diabetic nephropathy. Recent efforts at developing targetable transduction systems as vectors for gene delivery have made considerable progress toward addressing problems of cell type specific recognition and insertional mutagenesis. Whether cell-based strategies that incorporate RNA interference to silence disease-causing mutations can be applied in humans remains to be determined.
This article meets the Accreditation Council for Graduate Medical Education and the American Board of Medical Specialties Maintenance of Certification competencies for Patient Care and Medical Knowledge.
Footnotes
The authors have no financial or proprietary interest in the subject matter of this article.
- © Academic Division of Ochsner Clinic Foundation








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}