
Abstract
Background The most common lysosomal storage disorder, Gaucher disease, represents a collection of 3 clinical syndromes associated with disrupted glucocerebroside catabolism. Despite the common occurrence of dyspnea in advanced Gaucher, dyspnea is rarely reported as a presenting symptom of the disease.
Case Report A 10-month-old male was referred to the Otolaryngology Clinic for evaluation of progressive dyspnea. Physical examination was significant for cervical adenopathy, inspiratory stridor, and developmental delay. A complete evaluation for failure to thrive and lymphadenopathy was performed, with subsequent lymph node biopsy and enzyme assay confirming the presence of Gaucher disease.
Conclusion A high level of suspicion is required to make an early diagnosis of Gaucher disease, but it should be considered in patients presenting with failure to thrive, generalized lymphadenopathy, and respiratory or neurologic findings. Initiation of early treatment is paramount for the prevention of irreversible disease.
INTRODUCTION
Gaucher disease is an autosomal recessive lysosomal storage disorder caused by various mutations of the β-glucocerebrosidase gene (1q21). Mutation of this gene leads to functional impairment of the associated lysosomal hydrolase, with resultant accumulation of undigested glucocerebroside in histiocytes of the reticuloendothelial system. The accumulation of intracellular glucocerebroside leads to the formation of Gaucher cells, characterized by a foamy, glycolipid-laden cytoplasm with tubular formations of aggregated glucocerebroside. The most common lysosomal storage disorder, Gaucher disease includes 3 forms that are differentiated by age of onset and neurologic manifestations.1 Type 1 disease is the most common form, sparing the central nervous system and presenting throughout life with hepatosplenomegaly and bony lesions. Type 3 disease is intermediate in both prevalence and manifestations, presenting in early adolescence with neurologic symptoms such as spasticity and dementia.2
Type 2 Gaucher disease is the rarest and most severe form, occurring in 1/100,000 to 1/500,000 live births without ethnic predisposition.3 This acute, neuropathic form presents in both perinatal and infantile groups, with symptoms notable by 6 months of age. The clinical course of type 2 Gaucher disease is invariable despite the time of presentation and is characterized by rapidly progressive neurologic degeneration and organ failure. The average lifespan of an infant with type 2 Gaucher disease is 9 months. Findings at presentation include increased tone and oculomotor abnormalities such as strabismus and saccade initiation failure. Organomegaly, laryngospasm, and respiratory failure subsequently develop. Respiratory impairment is recognized as a late manifestation of Gaucher disease, with rare reports of patients initially presenting with signs of respiratory distress.3
CASE REPORT
An African-American male was born at term via caesarean section to a 31-year-old primigravid mother. The child's birth weight was 3,345 g. The father had reactive airway disease as a child, but the infant had no family history of neurologic, skeletal, or lymphoproliferative disorders. Despite minimal prenatal care, the child had no neonatal complications, passed his newborn hearing and metabolic screens, and was discharged home in good condition from the well-baby nursery.
The patient met all developmental milestones until 5 months of age, when he developed chronic cough and congestion associated with weakness, gastroesophageal reflux, and a single seizure witnessed by the patient's mother. The child's symptoms progressed despite outpatient antibiotic and reflux therapies. By 10 months, he was referred for evaluation of progressive dysphagia, stridor, and possible hearing loss.
At presentation, the infant's weight had decreased from the 50th to the 5th percentile for his age, and he displayed obvious developmental delay with neck arching and eye rolling in response to stimulation. He did not crawl, pull, or stand and did not hold a bottle. Cervical examination revealed extensive bilateral adenopathy with occasional inspiratory stridor. Lymphadenopathy was also palpated in the axillary and inguinal lymph nodes. He was tachypneic with coarse respiratory sounds bilaterally. An abdominal examination showed severe hepatosplenomegaly.
Flexible direct laryngoscopy of the upper airway revealed paradoxical movement of the vocal cords with frequent inspiratory adduction and abduction occurring every 3 breaths. The patient also had significant pooling of secretions and mild laryngomalacia.
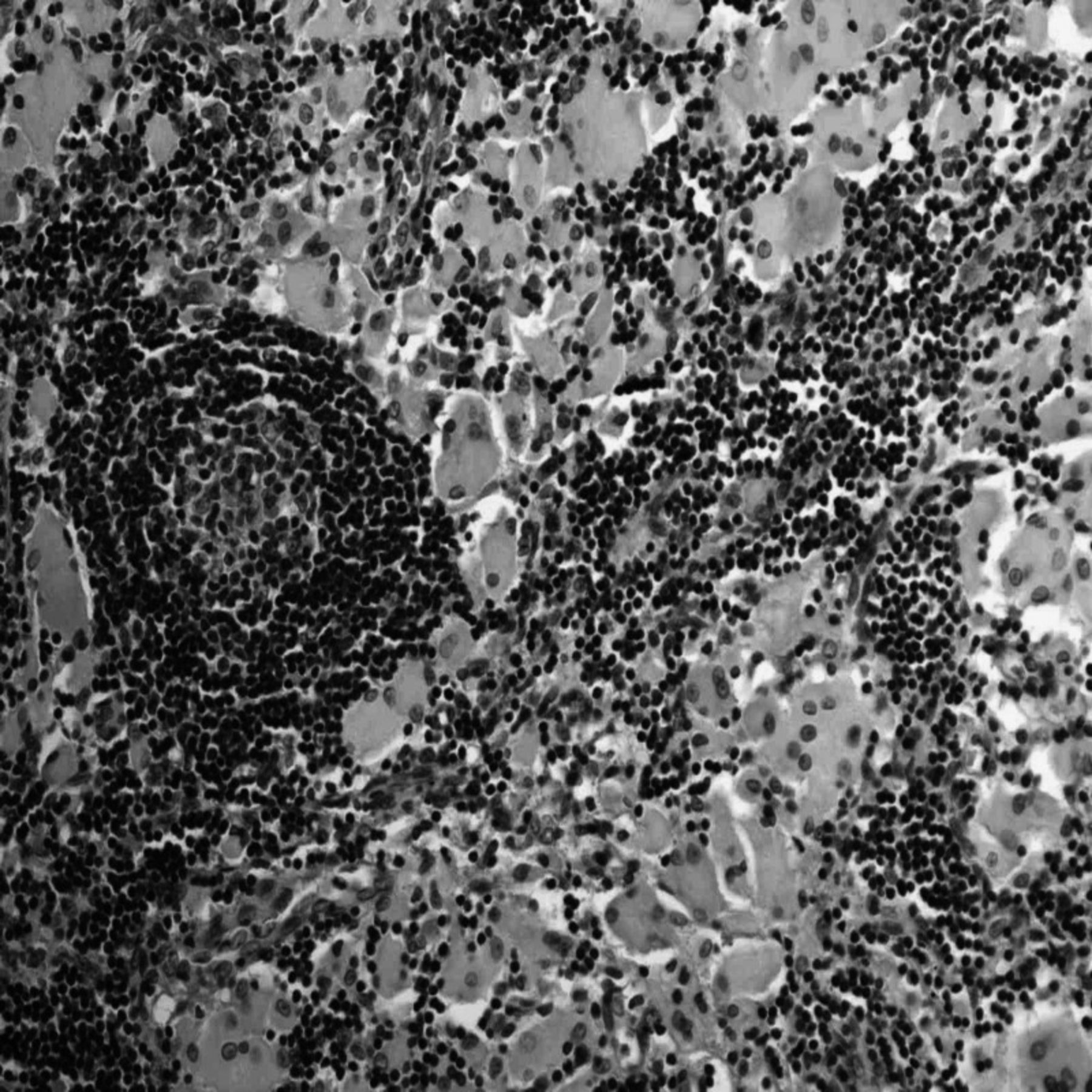
On subsequent admission to the pediatric intensive care unit, the patient was electively intubated. A complete evaluation for failure to thrive and lymphadenopathy was undertaken, and computed tomography revealed compressive bilateral cervical and mediastinal adenopathy extending from the mandible to the carina (Figure 1). Serum cultures ruled out infectious causes, with assays negative for hepatitis (B surface antigen/core antibody/autoantibodies), Epstein-Barr virus (immunoglobulin M/immunoglobulin G), Toxoplasma gondii (DNA polymerase chain reaction), HIV (enzyme-linked immunosorbent assay and polymerase chain reaction), and tuberculosis (purified protein derivative). Complete blood count was normal. Lymph node biopsy showed sinuses filled with large histiocytes that had frothy, wrinkled cytoplasms. B cells stained negative for CD21 and S100 and positive for KP-1 (CD-68) (Figure 2). We found no evidence of Langerhans cell histiocytosis, leukemia, or lymphoma. Serum amino acids were within normal limits, but a serum lysosomal enzyme assay revealed decreased leukocyte glucocerebrosidase activity, consistent with a diagnosis of Gaucher disease.

Computed tomography displaying compressive adenopathy extending from the level of the larynx to the mediastinum.

Hematoxylin and eosin lymph node biopsy showing collections of eosinophilic Gaucher cells with nearby lymphoid germinal center (40× magnification).
The patient remained intubated throughout the majority of his hospital stay because several attempts at extubation failed. Despite maximal conventional ventilator settings, he had persistent hypoxia. On hospital day 16, the family decided to withdraw life support.
DISCUSSION
The cervical lymphadenopathy, failure to thrive, and respiratory distress in this case are associated with several neonatal conditions, including infection, malignancy, and lysosomal storage disease. A high level of clinical suspicion is required to quickly identify a patient with Gaucher disease. While cytopenias, systemic lymphadenopathy, and hepatosplenomegaly are common manifestations of the disease, the low levels of serum leukocyte glucocerebrosidase activity specifically identify Gaucher, with activity between 0% and 30% of normal values confirming disease.4 This case illustrates that early diagnosis is paramount because the efficacy of enzymatic replacement and substrate reduction therapies decreases with the accumulation of lysosomal disease.
Enzymatic replacement therapy involves administration of recombinant human β-glucocerebrosidase to replace the impaired lysosomal enzyme in patients with Gaucher disease. This enzyme—sold as imiglucerase (Genzyme Therapeutics, Cambridge, MA)—is engineered to display a high level of lysosomal uptake but is unable to cross the blood-brain barrier. The efficacy of enzymatic replacement therapy is therefore limited to the treatment of symptomatic type 1 and visceral type 3 Gaucher disease and has not been approved for treatment of the mostly neuropathic type 2 form of the disease.5
Substrate reduction therapy seeks to prevent the accumulation of lysosomal glucocerebroside by inhibiting glucosylceramide synthase, the first step in glucocerebroside anabolism. Current treatment is with miglustat (Actelion, Basel, Switzerland)—a synthetic iminosugar with a large volume of distribution—to prevent the accumulation of glucocerebroside in lysosomes and macrophages of both the reticuloendothelial and central nervous systems. Recent studies have shown decreased hepatosplenomegaly in patients with type 1 Gaucher receiving oral therapy, and future studies are evaluating substrate reduction therapy in the treatment of patients with neuropathic forms of the disease.6,7
CONCLUSION
Type 2 Gaucher disease is a rare disorder of glucocerebroside metabolism, presenting in the first months of life with lymphadenopathy and progressive neurologic decline. A diagnosis of Gaucher disease should be considered when failure to thrive is associated with generalized lymphadenopathy and respiratory or neurologic findings. Although no treatment for the neurologic manifestations of type 2 Gaucher currently exists, substrate reduction therapy is a recently developed intervention with the potential to prevent both visceral and neuronal manifestations of disease.
This article meets the Accreditation Council for Graduate Medical Education and the American Board of Medical Specialties Maintenance of Certification competencies for Patient Care and Medical Knowledge.
Footnotes
The authors have no financial or proprietary interest in the subject matter of this article.
Presented at the SENTAC 2010 Annual Meeting, December 2-5, Cincinnati, OH.
- © Academic Division of Ochsner Clinic Foundation








{kind=link}
{kind=link}