
Abstract
Background Nitrates in drinking water are generally considered the sole source of nitrite poisoning with methemoglobinemia in infantile methomoglobinemia (IM). However, IM, which occurs during the first 4 months of life, is actually a constellation of cyanosis and hypoxia associated with methemoglobinemia that can result from several other causes.
Methods This review reexamines the role of nitrate levels in drinking water as a cause of IM and identifies other sources of nitrates that can affect public health and cause chronic diseases.
Results Causes of IM include nitrites in foods, environmental chemical exposures, commonly prescribed pharmaceuticals, and the endogenous generation of oxides of nitrogen. Infants with congenital enzyme deficiencies in glucose-6-phosphate dehydrogenase and methemoglobin reductase are at greater risk of nitrite-induced methemoglobinemia from nitrates in water and food and from exposures to hemoglobin oxidizers.
Conclusion Early epidemiological studies demonstrated significant associations between high groundwater nitrate levels and elevated methemoglobin levels in infants fed drinking water–diluted formulas. However, more recent epidemiological investigations suggest other sources of nitrogenous substance exposures in infants, including protein-based formulas and foods and the production of nitrate precursors (nitric acid) by bacterial action in the infant gut in response to inflammation and infection.
- Congenital methemoglobinemia
- drinking water
- glucose-6-phosphate dehydrogenase deficiency
- nitrates
- nitrites
INTRODUCTION
Nitrates in drinking water have long been considered the source of nitrite poisoning with methemoglobinemia in blue baby syndrome, also known as infantile methemoglobinemia (IM) to avoid confusion with cyanotic congenital heart diseases. IM is a constellation of cyanosis and hypoxia associated with methemoglobinemia occurring in the first 4 months of life. Clinical manifestations are directly proportional to blood methemoglobin levels. Levels of 10%-15% cause weakness and mild cyanosis; increasing levels cause irritability and tachyarrhythmias, with fatal cardiorespiratory collapse as levels surpass 70%.1-7
Early epidemiological studies demonstrated significant associations between high groundwater nitrate levels and elevated methemoglobin levels in infants fed drinking water–diluted formulas.1-7 However, recent epidemiological investigations suggest other sources of nitrogenous substance exposures in infants, including protein-based formulas and foods and the production of nitrate precursors (nitric acid) by bacterial action in the infant gut in response to inflammation and infection.7,8
This review reexamines the role of nitrate levels in drinking water as a cause of IM and identifies other sources of nitrates that can affect public health and cause chronic diseases such as hypertension and cancer.
METHODS
We queried internet search engines MEDLINE, PubMed, Ovid, Google, and Google Scholar with the keywords congenital methemoglobinemia, drinking water, glucose-6-phosphate dehydrogenase (G6PD) deficiency, nitrates, and nitrites to reexamine the role of nitrate levels in drinking water as the cause of IM and to identify other exogenous sources of nitrates that can affect the health of infants and adults alike and cause clinical manifestations other than methemoglobinemia.
NITROGEN CYCLE
Nitrates and nitrites are chemical compounds that are produced naturally in the nitrogen cycle and have a variety of industrial applications as food preservatives (sodium nitrite), explosives (nitrates), pesticides (sodium and potassium nitrate), fertilizers (ammonium nitrate), and fumigants (nitrates).1,2 In the nitrogen cycle, nitrogen sources from the atmosphere, animal and human wastes, decaying vegetation, and nitrate-containing fertilizers are initially converted to ammonia by saprophytic soil-dwelling bacterial action in a process known as ammonification (Figure 1).1-3 Ammonia is subsequently converted to nitrite by soil-dwelling, nitrogen-fixing bacteria in a process known as nitrification.1,2 Nitrites may be further oxidized by soil-dwelling nitrifying bacteria and enter groundwater as nitrates.1,2 In addition to assimilation into groundwater, nitrates are concentrated in surface and subsurface ground-dwelling plants and vegetables, such as carrots.1,2

The nitrogen cycle. (Reprinted from the United States Geological Survey.3)
GEOLOGICAL, AGRICULTURAL, AND SEASONAL INFLUENCES ON DRINKING-WATER NITRATE LEVELS
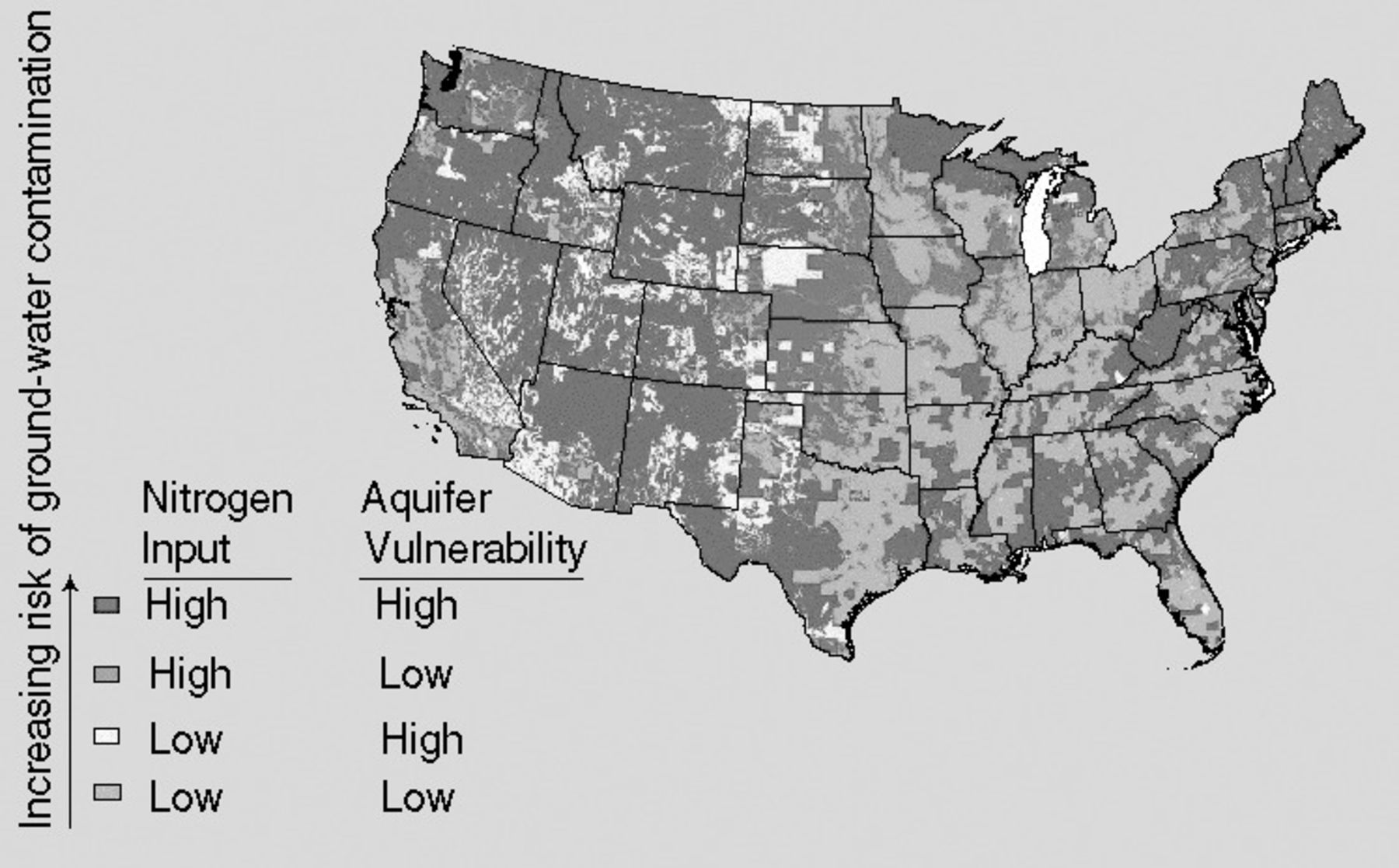
Nitrate levels in groundwater vary with geology, soil types, agricultural practices, and seasons across the United States (US). Groundwater exists at 2 subsurface levels: above (shallow groundwater) and below (deep groundwater) a depth of 5 meters.2 Shallow groundwater is more susceptible than deep groundwater to contamination by chemicals such as nitrogenous fertilizers applied to land surfaces. More than 50% of the US population receives drinking water from groundwater sources, such as domestic wells that access shallow groundwater or deep groundwater in major aquifers.2,4 The remainder of the US population receives drinking water from surface water sources, such as rivers and lakes.4 Although drinking water from aquifers and surface water sources is monitored for nitrate load, domestic wells, most of which are privately owned, are not continuously monitored (Figure 2) although the US Geological Survey National Water-Quality Assessment Program periodically monitors domestic wells for nitrate load.5,6 Of the privately owned domestic wells sampled for nitrates during the period 1993-2000, 9% exceeded the maximum contaminant level (MCL) of 10 mg/L nitrate, compared to only 2% for publicly owned groundwater access wells.6

United States regions of high nitrogen input contaminated groundwater and aquifer vulnerability to groundwater contamination by nitrates. (Reprinted from the United States Geological Survey.5)
Besides depth, other geological factors that can affect groundwater nitrate levels include soil porosity, soil drainage, and soil leachate penetration and accumulation.5,6 Porous and well-drained sandy and gravel-rocky soils allow more nitrites to enter groundwater, while nonporous clay soils provide a barrier to greater nitrogen loads, especially from agricultural practices such as crop cutting, crop burning, and animal manure and fertilizer applications.5,6 Other sources of surface nitrogen loads that can increase nitrate concentrations in sandy soils and shallow wells include atmospheric pollution from fossil fuel use in transportation, industry, and electrical power generation; leaking septic tanks; and improperly maintained septic drainage fields.4-6 Lastly, seasonal nitrogen contributions to drinking-water nitrate levels, especially in domestic well and aquifer-supplied drinking water systems, include increased nitric acid levels during heavy rainfalls (acid rain) and fluctuating water table depths in spring and fall.2,4-6
Many major US cities located along the Mississippi River and its tributaries, such as the Missouri and Ohio rivers, rely on river-treated water for metropolitan drinking water needs.4 Environmental studies have demonstrated minimal progress in reducing river nitrate levels since 1980 as a result of nitrate contributions from fluctuating water tables and from agricultural manure and fertilizer leachates entering shallow groundwater.4,5 Periodic nitrate level monitoring at 5 sites (1 on the Missouri River and the remaining 4 along the Mississippi River) have demonstrated 9%-97% increases in nitrate levels between 1980 and the present.4 In response, the Environmental Protection Agency (EPA) recommends that chronic oral exposure for infants should not exceed a nitrate level of 1.6 mg/kg/d and a nitrite level of 0.1 mg/kg/d.1 US drinking water advisories for a 4 kg child include an MCL goal of no more than 10 mg/L of nitrate and 1 mg/L of nitrite.1
The fluctuating levels of nitrates (nitrate flux) in the Mississippi River are primarily responsible for the size of the hypoxic or dead zone in the Gulf of Mexico off the Louisiana Coast, west of the Mississippi River Delta.4 The Gulf hypoxic zone is among the largest such zones in the world, and its size is determined by climatic and seasonal nitrate contributions from the Mississippi River.4 Climatic contributions include heavy precipitation in the southern Mississippi River drainage basin from late summer tropical storms and hurricanes.4 Seasonal contributions include snow melt and heavy spring rains that swell the river tributaries and deliver larger nitrate loads offshore in the spring than during the summer. The EPA has determined that spring snowmelt in the northern river basin and heavy spring rainfall in the southern basin deliver up to 50% of the annual nitrate load to the Mississippi River and are strong independent predictors of the size of the summertime Gulf hypoxic zone.4 Unless tropical storms and hurricanes intervene, summers are characterized by lower rainfall, higher temperatures, and lower river levels.4
EPIDEMIOLOGY OF NITRATE-INDUCED METHEMOGLOBINEMIA
Nitrates in drinking water are widely considered the sole source of nitrite poisoning with methemoglobinemia in IM. However, IM, which occurs during the first 4 months of life, is actually a constellation of cyanosis and hypoxia associated with methemoglobinemia that results from several other causes, including nitrites in foods (especially vegetables such as carrots), environmental chemical exposures, commonly prescribed pharmaceuticals, and even the endogenous generation of oxides of nitrogen.7 For example, nitric oxide is generated in the infant gut by the action of intestinal bacterial flora on nitrites in foods, especially during acute gastrointestinal illnesses characterized by inflammation or infection.7
When the iron in heme is reduced from its ferrous to its ferric state, hemoglobin is converted to methemoglobin, a form of hemoglobin that is unable to deliver oxygen to the tissues that become hypoxemic and then acidotic. When the proportion of methemoglobin reaches approximately 15% of the total circulating hemoglobin level, clinically detectable methemoglobinemia results with noticeable cyanosis and subsequent acidosis and cardiovascular depression. Because the most important oxidative stressors on hemoglobin are the nitrites produced by digestion of dietary nitrates and the oxygenation of hemoglobin is a minor oxidizing process, a small proportion of circulating hemoglobin is normally methemoglobin, usually 1%-3%.7 The most important chemoprotective mechanism against increasing methemoglobin levels in the circulation is the key enzyme methemoglobin reductase (NADH cytochrome b5 reductase) that can convert the ferric heme iron in methemoglobin back to the ferrous heme of hemoglobin capable of carrying oxygen.
The greatest risk factors for methemoglobinemia include (1) genetic deficiencies in key enzyme systems, such as methemoglobin reductase and G6PD, a red blood cell oxidant protector; (2) genetic abnormalities in hemoglobin structure and iron-carrying capacity; and (3) exposures to oxidizing chemicals, drugs, and, rarely, combinations of high nitrate foods.
Infants under 6 months of age, especially between birth and 4 months of age, are predisposed to methemoglobinemia because their immature hepatic cytochrome P450 system cannot produce adequate circulating levels of methemoglobin reductase. G6PD deficiency is the most common genetic enzyme deficiency and is especially prevalent in the US, affecting approximately 10% of African Americans and Americans of southern Mediterranean descent. G6PD is a critical enzyme in the synthesis of reduced nicotinamide dinucleotide phosphate (NADPH), which activates the enzyme NADPH-dependent methemoglobin reductase to rapidly reduce methemoglobin levels exceeding normal range (1%-3%).7,8
In addition to high nitrate levels in well-supplied drinking water, other oxidative stresses that infants may face include high nitrate levels in foods, such as processed baby foods (especially vegetables) and sodium nitrite–treated meats (sausage, bacon, and bologna), and the endogenous production of nitrites from nitric oxide generation. However, even though >90% of dietary nitrates come from foods, only 1 reported case of IM was caused by food, specifically overdoses of carrot juice that contained >700 times the MCL for nitrites (775 ppm).8
For decades, IM was blamed solely on nitrates in the drinking water used to prepare infant formulas, but research has implicated genetic enzyme deficiencies, inheritable hemoglobinopathies, and periodic generation of nitric oxide levels high enough to overpower protective methemoglobin-reducing systems during acute gastrointestinal infections as the most common causes of IM.7 Because research refutes exogenous nitrate-to-nitrite sources as causes of IM and supports endogenous nitrite production secondary to genetic abnormalities or nitric oxide generation in an inflamed infant gut as causative mechanisms for IM, perhaps the current MCL for nitrate in drinking water of 10 mg/L (or 10 ppm) should be revisited.7
Nevertheless, persistent high dietary nitrate levels and congenital inabilities to handle normal nitrate loads may be detrimental and result in the formation of carcinogenic N-nitroso compounds.1 Although the data are inconsistent and do not currently support causation, high dietary nitrate loads have been correlated with carcinomas, hypertension, and thyroid dysfunction in adults and with brain tumors, nasopharyngeal tumors, and leukemia in children.1
RISK FACTORS AND EFFECT MODIFIERS FOR METHEMOGLOBINEMIA
In addition to the congenital inability to handle nitrate loads in infancy because of key enzyme deficiencies, methemoglobinemia may result from accidental and therapeutic exposures to chemicals, especially aniline dyes, and several medications, such as nitrite-based drugs, including nitroglycerin and sodium nitroprusside; the local anesthetics benzocaine and prilocaine; the antimalarials chloroquine and primaquine; sulfonamide antibiotics, including dapsone and trimethoprim-sulfamethoxazole; and a few miscellaneous medications, such as phenazopyridine hydrochloride (Pyridium) and metoclopramide (Reglan).9 The local anesthetics are an important class of oxidizing drugs, especially the commonly used, topically applied benzocaine and prilocaine that are often used in analgesic mouthwashes and ointments for children.9 All of these chemicals and pharmaceuticals can oxidize hemoglobin and cause methemoglobinemia, which if untreated above levels of 10%-15% may result in confusion, cyanosis, imbalance, hemodynamic instability, coma, and death.9
The congenital hemoglobinopathy HbM is characterized by the formation of abnormal hemoglobin with an aberrant histidine residue near the heme ring that stabilizes heme iron in the ferric or reduced state and limits oxygen transport.9 HbM heterozygotes are generally asymptomatic, but patients may suffer fulminant hemolytic crisis after receiving oxidant drugs.9 Therefore, prilocaine, nitroprusside, or other oxidant drugs should be avoided in any identified HbM phenotype.9 Drugs that form peroxides by interactions with oxyhemoglobin may also trigger hemolysis in oxidizer-exposed patients.9 Lastly, chronic hemolytic anemias may complicate acute methemoglobinemia from oxidizing chemical and drug exposures in children and adults with congenital key enzyme deficiencies and hemoglobinopathies.9
PATHOPHYSIOLOGY OF METHEMOGLOBINEMIA
In IM, nitrates bind to fetal hemoglobin and result in the production of methemoglobin by reducing the heme iron in the ferrous (+2) state to the ferric (+3) state. Fetal hemoglobin is the major hemoglobin component during intrauterine life. Its levels in circulating blood cells decrease rapidly during infancy and reach a concentration of 0.5% in adults. By 2 to 3 months of age, however, physiologic anemia results. After 3 months, progressive increases occur in erythrocyte mass and hematocrit. By 4 to 6 months, the oxyhemoglobin dissociation curve approximates that of adults. Characteristics of fetal hemoglobin influence oxygen transport. For example, fetal hemoglobin has a P50 value of 19 mmHg compared to 26 mmHg for adults, which results in a leftward shift of the fetal oxyhemoglobin dissociation curve. Subsequent increased affinity of hemoglobin for oxygen manifests as decreased oxygen release to peripheral tissues.9
Red blood cell hemolysis may occur in chronic, indolent cases of methemoglobinemia. Hemolytic crises may be induced through infections or ingestion of oxidizing agents. The outcomes of untreated hemolysis include progressive cyanosis with acidosis and potentially fatal cardiovascular depression. Besides hemolysis, other noteworthy complications of methemoglobinemia may be iatrogenic as a result of overtreatment with methylene blue.
DIAGNOSIS OF METHEMOGLOBINEMIA
The diagnosis of methemoglobinemia rests on clinical suspicion of methemoglobinemia as the cause of cyanosis, differential exclusion of other common causes of cyanosis, and selection of the most precise instruments to measure methemoglobin levels. A suspected diagnosis of methemoglobinemia is suggested by a combination of the following observations: (1) a prior history of cyanosis following oxidizing drug or toxic chemical exposures; (2) a low transcutaneous oxygen saturation (SpO2) in room air that hovers around 85% in 100% oxygen; (3) a discrepancy between low SpO2 on pulse oximetry and high arterial oxygen partial pressure (PaO2) and arterial oxygen saturation (SaO2) in arterial blood gases; and (4) dark or chocolate-colored arterial blood.10 A rapid differential diagnosis should include unrecognized intraanesthetic esophageal intubation, massive blood loss, myocardial depression, atelectasis, pulmonary embolism, carbon monoxide or hydrogen sulfide poisoning, congenital hemoglobinopathies (especially HbM), and key enzyme deficiencies (G6PD deficiency more commonly than hemoglobin reductase deficiency).
Selection of the right monitoring instruments to confirm a diagnosis of methemoglobinemia is critical because many otherwise reliable instruments will give false measurements, especially pulse oximetry and arterial blood gas analysis. Pulse oximetry detects the peak wavelengths of oxyhemoglobin (940 nm) and deoxyhemoglobin (66 nm), calculates their transformed ratio as SpO2, and falsely reads SpO2 during methemoglobinemia: underreading SpO2 at low methemoglobin levels and overreading SpO2 at high methemoglobin levels.10 Arterial blood gas analyzers measure gas pressures instead of wavelengths, extrapolate SaO2 from the measurement of PaO2, and also falsely read SaO2 during methemoglobinemia.10 Therefore, the only 2 instruments that can precisely confirm a diagnosis of methemoglobinemia are (1) transcutaneous CO-oximeters that can also rule out carbon monoxide and hydrogen sulfide poisoning and precisely measure carboxyhemoglobin, methemoglobin, and sulfmethemoglobin levels, and (2) more sophisticated (and more expensive) transcutaneous spectrophotographic analyzers for oxyhemoglobin, deoxyhemoglobin, and methemoglobin.
MANAGEMENT OF METHEMOGLOBINEMIA
The general management of methemoglobinemia includes a combination of (1) identifying and discontinuing exposures to the offending oxidant drug or chemical, if present; (2) hyperoxygenation with 100% O2 or hyperbaric O2 in severe cases; (3) correcting any preexisting severe anemia and restoring hemoglobin and hematocrit with packed red blood cell transfusions, with exchange transfusions reserved for severe cases; (4) correcting acidosis and restoring arterial blood pH to 7.4 with intravenous aliquots of sodium bicarbonate; and (5) improving tissue perfusion with intravenous volume loading and vasopressors as indicated.10,11
Specific pharmacologic therapy for methemoglobinemia, unless contraindicated, is the intravenous administration of the antidote methylene blue. Methylene blue is an aromatic dye and an oxidizing agent, formerly used as an antimalarial and used today as an intravenous indicator dye for urine extravasation. It turns the urine green and the sclerae blue. Methylene blue therapy is indicated for methemoglobin levels ≥20% but is contraindicated in G6PD deficiency because of increased red blood cell hemolysis and is contraindicated in methemoglobin reductase deficiency because of refractoriness and risk of worsening methemoglobinemia. Methylene blue should be administered intravenously as a 1% solution at a dose of 1-2 mL/kg (1-2 mg/kg) to reduce the methemoglobin level to ≤10%.9,10 The same dose may be repeated intravenously in 30-60 minute increments to a maximum daily dose of 4-15 mL/kg/d (4-15 mg/kg/d), as doses greater than 15 mL/kg/d have induced methemoglobinemia.9,10 The major adverse effects of methylene blue therapy include nausea, vomiting, diarrhea, dyspnea, diaphoresis, and, rarely, anaphylactoid reactions.
Although not supported by randomized controlled investigations, adjunctive antioxidant therapy in refractory cases of methemoglobinemia or in cases in which methylene blue is contraindicated includes the intravenous administration of antioxidant vitamins such as ascorbic acid (vitamin C, 600 mg intravenously in normal saline solution).9,10 In cases where methylene blue therapy is contraindicated, such as prior anaphylactoid reaction, methemoglobin reductase deficiency, or G6PD deficiency, the general management steps of intravenous fluid loading, vasopressor reversal of hypotension as indicated, and correction of metabolic acidosis for methemoglobin levels ≥20% should be instituted with considerations for packed red blood cell or total exchange transfusions at methemoblobin levels <20%.
PREVENTION OF METHEMOGLOBINEMIA
Evidence suggests that limiting nitrate exposure in infants less than 6 months of age through drinking-water restrictions may be ineffective given high nitrate and nitrite loads in some foods and the production of the potent oxidizer nitric oxide in the infant gastrointestinal tract during acute gastrointestinal infections.12 Nevertheless, breastfeeding or substituting bottled water for high-nitrate-level drinking water from wells and aquifers for formula mixing is recommended in regions with high levels of nitrates, especially for infants with known G6PD deficiency, methemoglobin reductase deficiency, or HbM hemoglobinopathy.12 Recognized oxidant drugs should also be avoided in infants less than 6 months of age if possible, especially in infants with risk factors for methemoglobinemia.
Parents and family members should also be educated on the signs and symptoms of complications caused by methemoglobinemia in the event that the child experiences toxicity. Likewise, newborn screening for hereditary G6PD deficiency should be considered in the specific regions, particularly along the Mississippi River basin, with the highest levels of nitrates in well and surface drinking water.
CONCLUSIONS
Early epidemiological studies demonstrated significant associations between high groundwater nitrate levels and elevated methemoglobin levels in infants fed drinking water–diluted formulas. However, recent epidemiological investigations have suggested other sources of nitrogenous substance exposures in infants, including protein-based formulas and foods and the production of nitrate precursors (nitric acid) in the infant gut in response to inflammation and infection. The risk factors for infant methemoglobinemia are now well defined and are principally inheritable genetic deficiencies of key antioxidant enzyme systems and hemoglobin abnormalities. Methemoglobinemia should be suspected in infants with known risk factors or under therapy with known oxidant drugs. Methemoglobinemia should be treated in all cases with the recommended general management strategies and supplemented with methylene blue antidotal therapy unless contraindicated.
This article meets the Accreditation Council for Graduate Medical Education and the American Board of Medical Specialties Maintenance of Certification competencies for Patient Care and Medical Knowledge.
Footnotes
*: Dr Richard is now a resident in the Department of Anesthesiology at Beth Israel Hospital, Harvard Medical School, Boston, MA.
The authors have no financial or proprietary interest in the subject matter of this article.
- © Academic Division of Ochsner Clinic Foundation
In this issue








{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- INTRODUCTION
- METHODS
- NITROGEN CYCLE
- GEOLOGICAL, AGRICULTURAL, AND SEASONAL INFLUENCES ON DRINKING-WATER NITRATE LEVELS
- EPIDEMIOLOGY OF NITRATE-INDUCED METHEMOGLOBINEMIA
- RISK FACTORS AND EFFECT MODIFIERS FOR METHEMOGLOBINEMIA
- PATHOPHYSIOLOGY OF METHEMOGLOBINEMIA
- DIAGNOSIS OF METHEMOGLOBINEMIA
- MANAGEMENT OF METHEMOGLOBINEMIA
- PREVENTION OF METHEMOGLOBINEMIA
- CONCLUSIONS
- Footnotes
- REFERENCES
- Figures & Data
- References
- Info & Metrics