
Abstract
Background: Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome is a rare inherited disorder that results in waxing and waning nervous system and muscle dysfunction. MELAS syndrome may overlap with other neurologic disorders but shows distinctive imaging features.
Case Report: We present the case of a 28-year-old female with atypical stroke-like symptoms, a strong family history of stroke-like symptoms, and a relapsing-remitting course for several years. We discuss the imaging features distinctive to the case, the mechanism of the disease, typical presentation, imaging diagnosis, and disease management.
Conclusion: This case is a classic example of the relapse-remitting MELAS syndrome progression with episodic clinical flares and fluctuating patterns of stroke-like lesions on imaging. MELAS is an important diagnostic consideration when neuroimaging reveals a pattern of disappearing and relapsing cortical brain lesions that may occur in different areas of the brain and are not necessarily limited to discrete vascular territories. Future studies should investigate disease mechanisms at the cellular level and the value of advanced magnetic resonance imaging techniques for a targeted approach to therapy.
INTRODUCTION
Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome is a rare, inherited disorder that results in nervous system and muscle dysfunction and has distinctive imaging features. Most cases of MELAS syndrome are associated with a DNA point mutation, first described in 1990, that affects proteins critical to intracellular energy production.1 Neuroimaging reveals a characteristic pattern of disappearing and relapsing cortical brain lesions that may occur in different areas of the brain and are not necessarily limited to discrete vascular territories.
We present a rare case of MELAS syndrome and review the unique clinical, imaging, diagnostic, and management considerations of the disease.
CASE REPORT
A 28-year-old black female originally presented in 2011 with a week of headache and altered mental status. At that time, the family reported that she had had seizure-like activities during a recent imprisonment and that in response to questions, she gave inappropriate answers. Most of her serum laboratory values were within normal limits, but she had an elevated lactate level of 3.1 mmol/L (normal 0.5-1 mmol/L). Electroencephalogram (EEG) demonstrated beta activity with periodic high-amplitude delta waves and slow background activity localized to the left posterior temporal region consistent with an epileptogenic structural abnormality in this region, as well as widespread cerebral dysfunction. Magnetic resonance imaging (MRI) at that time demonstrated diffuse edema in the left temporal lobe with thickening of the meninges and mild dilation of the fourth ventricle. The working differential was herpetic meningoencephalitis, and although the patient tested negative for herpes simplex virus, she was treated with acyclovir. Her symptoms improved quickly, and after 3 days she was discharged on topiramate for seizure control.
She returned for outpatient follow-up and further testing in late 2011. She had an abnormal EEG after her original presentation showing evidence of epileptiform activity and structuring abnormality, mostly localized to the left hemisphere and frontotemporal area, with a mild degree of cerebral dysfunction. In late 2011, MRI showed that the left temporal area had resolved almost completely, but magnetic resonance spectroscopy (MRS) demonstrated elevated lactate peaks that were sufficiently high, in combination with the clinical findings, to be consistent with MELAS syndrome rather than with meningoencephalitis. She was started on levetiracetam, L-carnitine, and coenzyme Q10.
She was referred to our institution for evaluation of new-onset blurred vision and diplopia in the setting of chronic headaches, recurrent seizures, and transient stroke-like episodes. After a thorough physical examination and chart review, the patient was admitted. Computed tomography (CT) of the head at the referral presentation demonstrated left temporal hypoattenuation concerning for an acute vascular event, as well as slight atrophic changes and ventricular prominence that were atypical for the patient's age (Figure 1). MRI of the brain performed during the same admission demonstrated a corresponding T2-weighted fluid-attenuated inversion recovery (T2-FLAIR) hyperintense signal in the left temporal lobe, as well as primarily cortical diffusion weighted imaging (DWI) hyperintensity, although without apparent diffusion coefficient (ADC) abnormality (Figure 2). Based on the imaging findings, the differential was fairly broad and included encephalitis (viral, bacterial, and prion disease), vasculitis, ischemia (venous and embolic), and metabolic causes (MELAS syndrome, myoclonic epilepsy with ragged-red fibers, Leigh syndrome, and Kearns-Sayre syndrome). However, review of the patient's history narrowed the differential and revealed a longstanding history of MELAS syndrome, as well as a brother and child with mitochondrial disorders. Prior to this referral presentation, she had presented multiple times during acute exacerbations with muscle spasms, aphasia, hemianopsia, psychosis, and headache. Her symptoms quickly resolved, and she was discharged after 2 days of symptomatic management and monitoring.
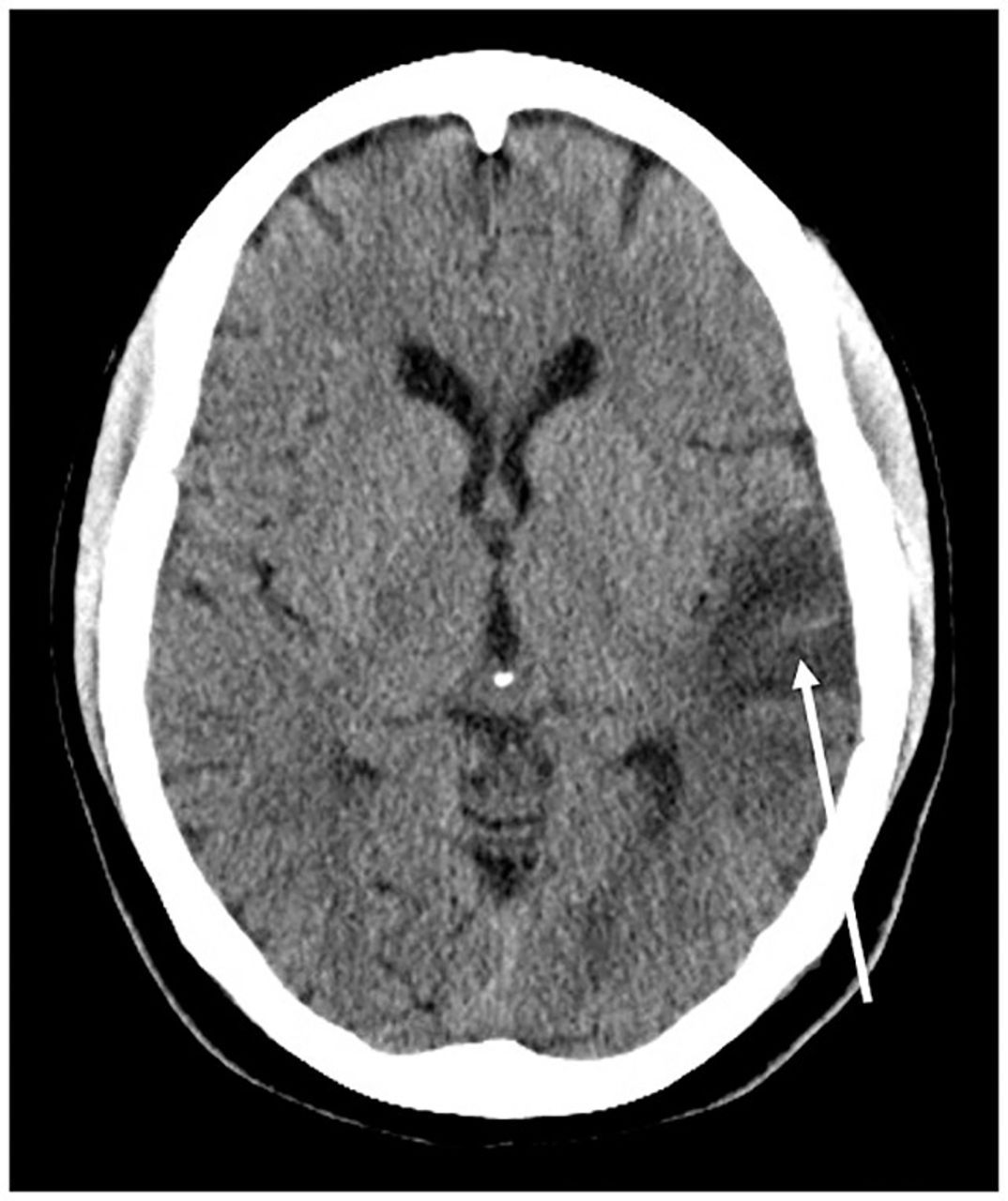
Computed tomography of the head without intravenous contrast at the patient's initial presentation demonstrates hypoattenuation and swelling in the left temporal lobe (arrow), as well as mild atrophy and ventricular prominence that are greater than expected for the patient's age of 28 years.
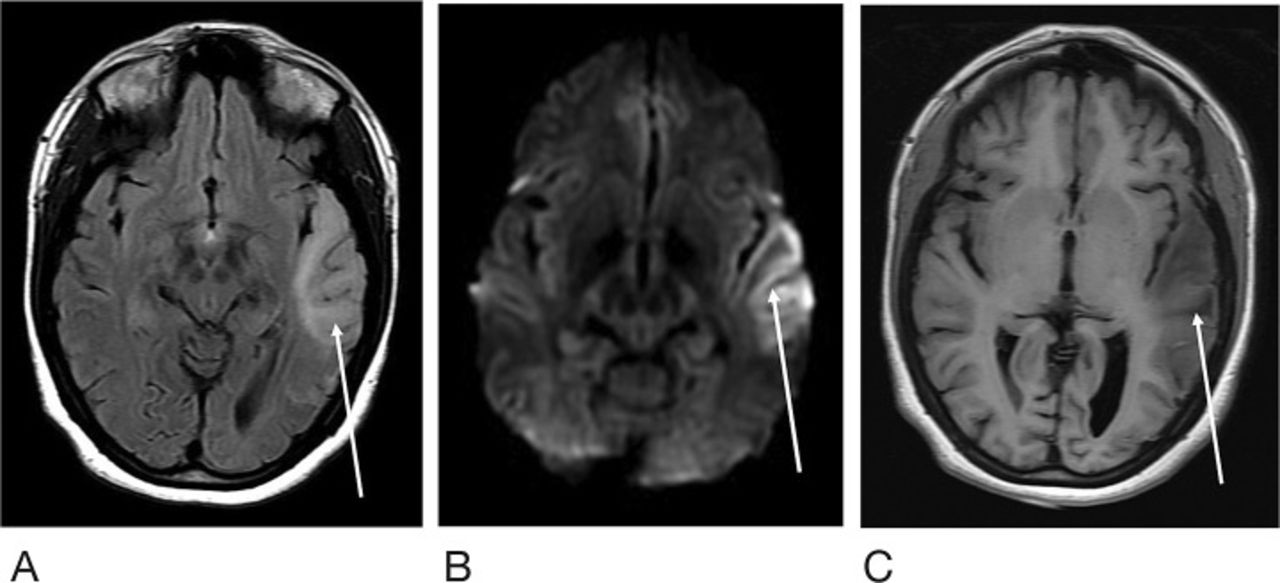
Magnetic resonance imaging of the brain at the level of the cerebral peduncles at the patient's initial presentation shows the left temporal lobe (arrow) demonstrating T2-weighted fluid-attenuated inversion recovery (T2-FLAIR) hyperintense signal (A), corresponding diffusion weighted imaging signal abnormality (B), and T1 hypointense signal (C). No corresponding apparent diffusion coefficient abnormality is apparent.
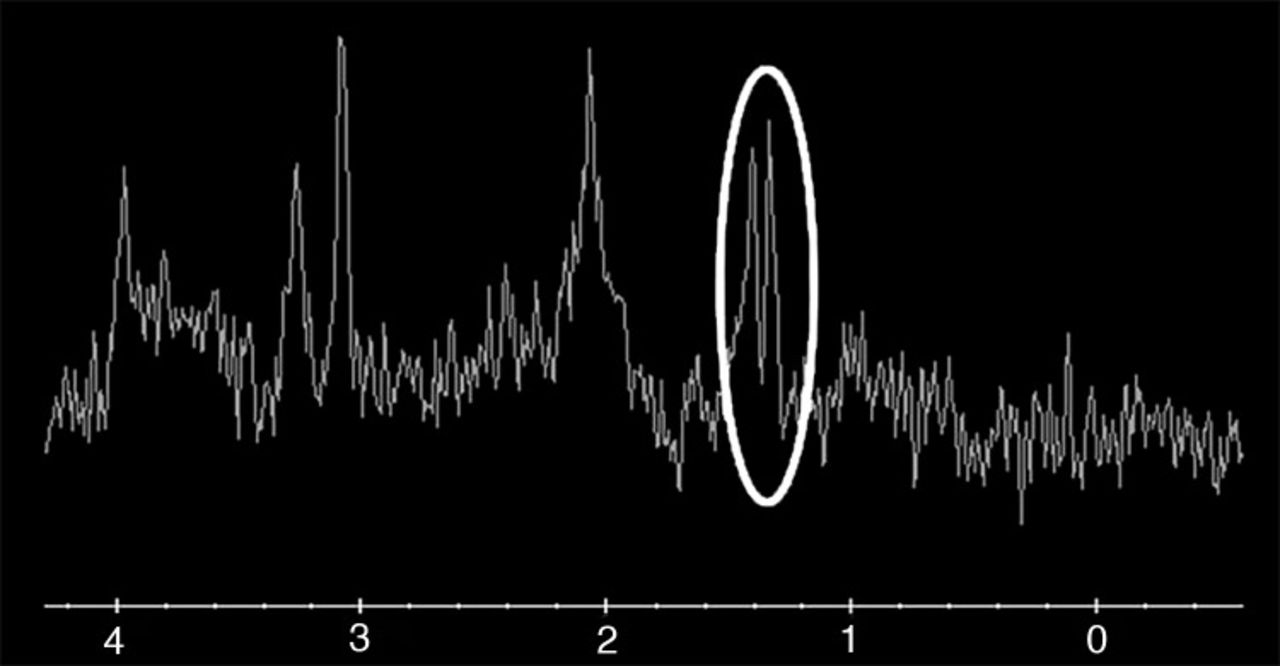
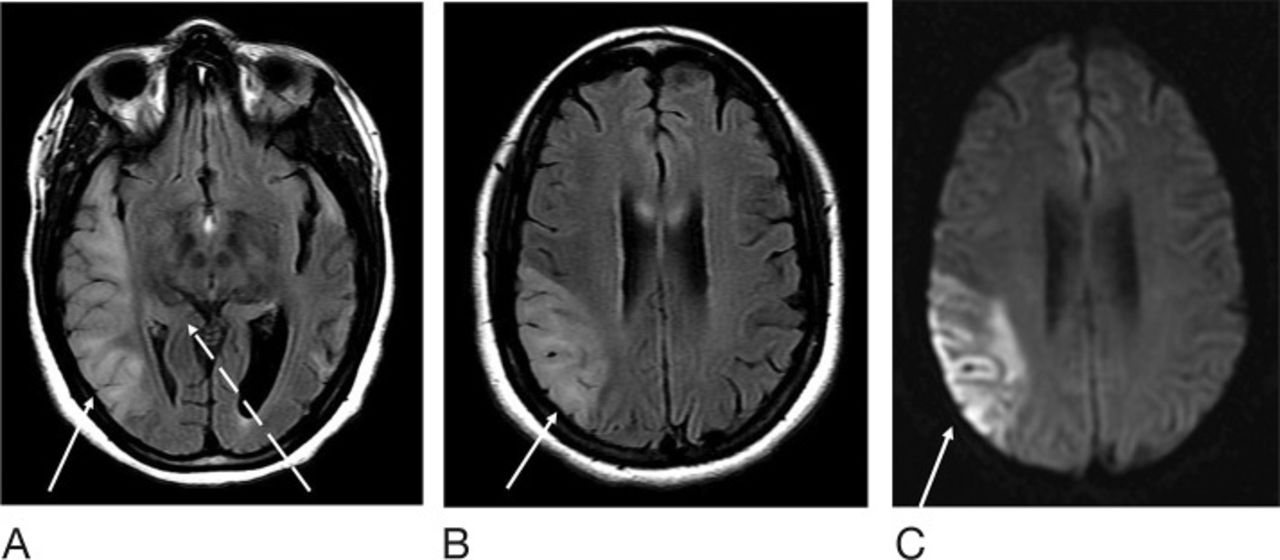
At the patient's emergency department (ED) visit 4 months after her initial referral, MRI of the brain demonstrated near-complete resolution of the left temporal T2-FLAIR hyperintense signal, with development of a new T2-FLAIR signal in multiple parts of the right temporal lobe, including the right temporal-occipital region. Corresponding DWI signal abnormality was not clearly present to a similar degree as her referral presentation (Figure 3). Functional MRS performed at 4 months exhibited a TE=35 ms lactate peak at 1.3 ppm, confirming defects in oxidative metabolism (Figure 4). CT of the head performed at another ED visit at 5 months showed both new and resolved hypoattenuation in similar distributions, as well as slight progression of ventricular prominence and atrophy (Figure 5). MRI of the brain 9 months after the patient's initial referral presentation showed mixed changes with resolution of the right temporal-occipital T2-FLAIR signal but progression that involved the majority of the right temporal lobe. The adjacent right parietal and occipital parenchymas were involved as well, with associated restricted diffusion in the subcortical white matter (Figure 6). Corresponding low-signal intensity on the ADC map (not shown) confirmed restricted diffusion. The patient has returned several more times with fluctuating levels of consciousness, recurrent migraines, and her hands and feet locking up. She was last seen in October 2016 for follow-up with complaints of migraines but no seizure or stroke-like activity. She continues to take levetiracetam, L-carnitine, and coenzyme Q10 for management of her condition.
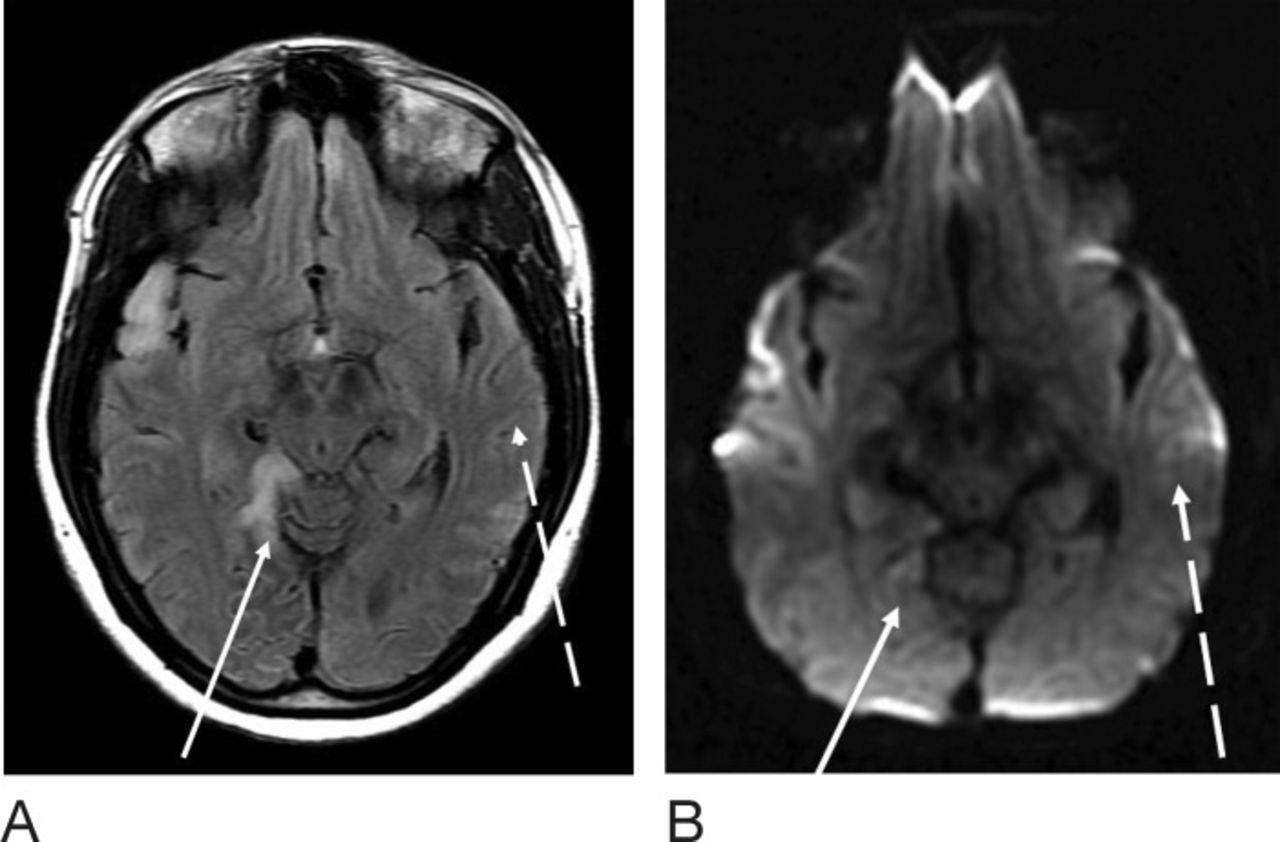
Magnetic resonance imaging of the brain at the level of the cerebral peduncles 4 months after the patient's initial presentation shows T2-weighted fluid-attenuated inversion recovery (T2-FLAIR) (A) and diffusion weighted imaging sequences (B). The left temporal T2-FLAIR hyperintense signal has almost completely resolved (dashed arrow); however, new abnormal signal is noted at multiple parts of the right temporal lobe anteriorly and the right temporal-occipital region (solid arrow). Very little diffusion weighted imaging and no apparent diffusion coefficient abnormality are apparent.

Magnetic resonance spectroscopy performed at the right temporal-occipital focus 4 months after initial presentation exhibited a TE=35 ms elevated lactate peak at 1.3 ppm (circled), indicating a defect in oxidative metabolism.

Computed tomography of the head 5 months after the patient's initial presentation at approximately the level of the third ventricle (for comparison with Figure 1) demonstrates interval resolution of the left temporal edema/hypoattenuation (dashed arrow) with new right inferior temporal hypoattenuation (solid arrow) and slight increased prominence of the ventricles.
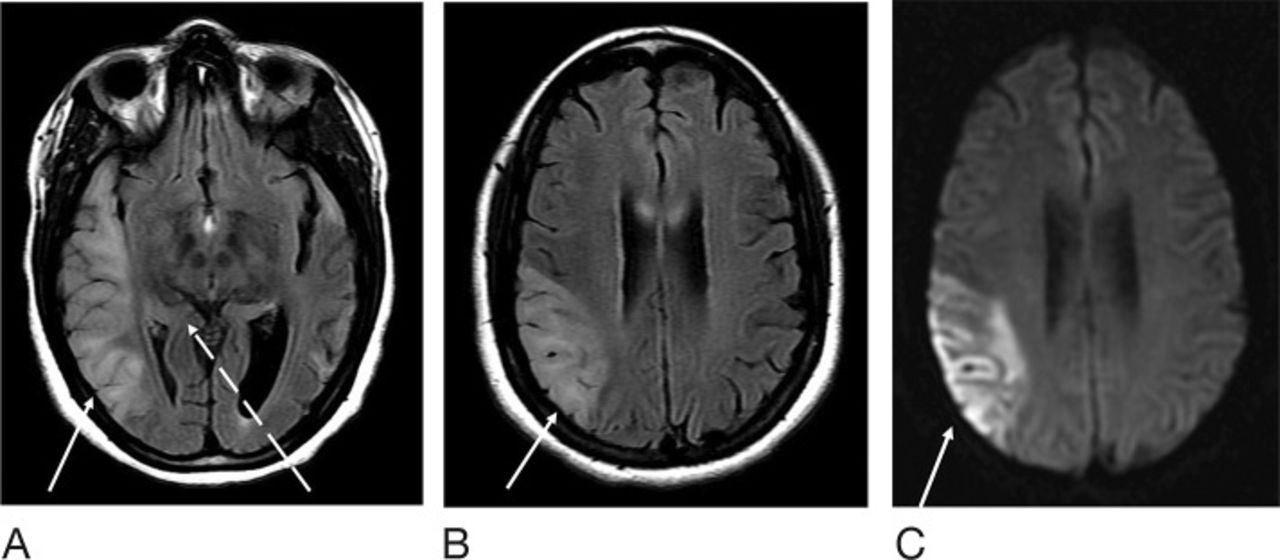
Magnetic resonance imaging of the brain 9 months after the patient's initial presentation at the level of the cerebral peduncles (for comparison with Figures 2 and 3). T2-weighted fluid-attenuated inversion recovery (T2-FLAIR) sequence demonstrates resolution of abnormal signal at the right temporal-occipital parenchyma (dashed arrow), with increased right temporal signal laterally (solid arrow) (A). The left temporal lobe is unchanged. T2-FLAIR (B) and diffusion weighted imaging (C) sequences at the level of the corpus callosum are also shown. T2-FLAIR hyperintense signal extends to the parietal lobe with restricted diffusion (solid arrow).
DISCUSSION
Overview
MELAS syndrome is a rare inherited disorder of intracellular energy production that typically presents prior to age 40. Eighty percent of cases are associated with the mitochondrial transfer RNA A3243G point mutation.1,2 As with other mitochondrial disorders, MELAS syndrome displays maternal inheritance with relative penetrance based on the degree of heteroplasmy. Thus, phenotype expression and severity of disease are related to the proportion of mutant DNA and residual activity of respiratory chain complexes in mitochondria within each cell. The disease activity, penetrance, and severity can be correlated to clinical presentation and characteristic imaging findings.3,4
Mechanisms of Disease
The literature proposes 3 potential mechanisms of disease pathology at the cellular level. The ischemic vascular theory states that an accumulation of dysfunctional mitochondria in microvasculature disrupts circulation and nitric oxide production by vascular endothelium, resulting in hypoperfusion and ultimately, widespread episodes of cerebral ischemia. The generalized cytopathic theory suggests that mutations alter oxidative phosphorylation at the cellular level, and defects in intracellular energy production produce clinical symptoms. The nonischemic neurovascular theory attributes stroke-like episodes to altered ion homeostasis and changes in vascular permeability, causing cellular hyperexcitability and cerebral blood flow changes.3,5
Presentation
MELAS syndrome targets organ systems with high metabolic activity, including the nervous and cardiovascular systems, and clinical onset typically occurs in early adulthood after a seemingly normal childhood with development attributable to cumulative effects of chronic lactic acidosis. As a result, focal neurologic deficits and psychiatric manifestations are well documented in this syndrome, and patients generally present prior to age 40 with a combination of migraine, seizures, altered mental status, ataxia, sensorineural hearing loss, and other focal deficits as a result of lactic acidosis and cerebral ischemia. The clinical effects are widespread, and the long-term sequelae may include progressive hearing loss, gait ataxia, psychiatric instability, glucose intolerance, and hypertrophic cardiomyopathy.4
Imaging and Diagnosis
Imaging of MELAS syndrome includes characteristic CT, MRI, and MRS findings in both the acute and chronic phase. Imaging in the acute phase typically reveals stroke-like cortical lesions that have a predilection for the parietal and occipital lobes, may span multiple cerebral vascular territories, and demonstrate a dynamic shifting spread: lesions classically appear, disappear, and reappear elsewhere over time.
Classically, noncontrast CT findings can appear normal, or noncontrast CT may show swelling in the acute phase and atrophy or lacunes if chronic (Figure 1). CT angiography may show early enhancement associated with hyperperfusion, and routine contrast-enhanced CT may demonstrate variable gyriform enhancement.
MRI is the gold standard for diagnosis and monitoring. MRI reveals global changes in gray-white differentiation, multifocal cortical and subcortical lesions that cross vascular territories, and varying degrees of generalized cerebral and cerebellar atrophy (Figures 2 and 3). Encephalomalacia may also be present in previously affected areas. The Table lists the differences noted on T1- and T2-weighted images in the acute and chronic settings. Diffusion-weighted MRI shows a cortical ribbon-like high-intensity signal consistent with diffusion restriction. During attacks, restricted diffusion may be present in the cortex, subcortical white matter, and basal ganglia.
Comparison of T1- and T2-Weighted Images Seen in Acute and Chronic Cases of Mitochondrial Encephalomyopathy With Lactic Acidosis and Stroke-Like Episodes (MELAS) Syndrome
Advanced MRI sequences can aid in the diagnosis of MELAS syndrome and provide additional insight into the mechanism of disease. Multivoxel MRS demonstrates a lactate peak in acutely abnormal brain regions associated with oxygen deficiency of cells in affected cortical areas.5 Several studies describe the potential value of nonanatomic MRI techniques—such as contrast-enhanced perfusion studies, blood-oxygen-level dependent imaging, oxygen extraction fraction, arterial spin labeling, and MRS—in identifying metabolic changes within lesion foci in the acute setting.5-8
Disease Management
MELAS syndrome disrupts the homeostasis of multiple metabolic pathways and requires an interdisciplinary team to treat the neurologic, endocrine, cardiac, and psychiatric manifestations of the disease. While conventional pharmacologic agents are used for symptomatic support, commonplace medications that may exacerbate the etiology of the disease must be avoided. For example, while anticonvulsants are effective for seizure prevention, valproic acid may paradoxically lower the seizure threshold in patients with MELAS syndrome because of mitochondrial injury and should therefore be avoided. Additionally, standard hypoglycemic agents may treat glucose intolerance; however, metformin should be avoided because of its superimposed risk for lactic acidosis in MELAS syndrome.9
Despite limited research-based evidence, supplementation with a combination of antioxidant and cofactor therapies is common practice and seeks to replenish constituents in oxidative metabolism pathways. Several studies have shown that L-arginine and citrulline may decrease the frequency and morbidity of stroke-like episodes because of an increase in nitric oxide bioavailability.10,11 Coenzyme Q10 may improve the efficacy of the electron transport chain through its antioxidant effects.9 Additional studies have proposed a potential benefit of creatine monohydrate and L-carnitine to enhance metabolism in mitochondrial disease.9
CONCLUSION
MELAS is a rare syndrome caused by genetic defects in mitochondrial proteins involved in oxidative phosphorylation. This case presents a classic example of the relapse-remitting disease progression with episodic clinical flares and fluctuating patterns of stroke-like lesions on imaging. Future studies should investigate disease mechanisms at the cellular level and the value of advanced MRI techniques for a targeted approach to therapy.
This article meets the Accreditation Council for Graduate Medical Education and the American Board of Medical Specialties Maintenance of Certification competencies for Patient Care and Medical Knowledge.
ACKNOWLEDGMENTS
The authors have no financial or proprietary interest in the subject matter of this article.
- © Academic Division of Ochsner Clinic Foundation 2017








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}