
Abstract
Background: Heterotaxy is a condition of abnormal lateralization of organs across the body’s left-right axis, causing multiple congenital malformations. The anatomic manifestations of heterotaxy syndrome generally follow one of two patterns, referred to as right atrial isomerism (with two similar right atria and duplication of right-sided features of multiple organs) and left atrial isomerism (with two similar left atria and duplication of left-sided features of multiple organs). Cardiac surgical intervention for patients with heterotaxy syndrome depends on ventricular physiology and circulatory balance. For patients with single-ventricle physiology, a Fontan operation, which directs systemic venous return to the pulmonary arteries, is the definitive intervention. Prior to a Fontan operation, many patients require one or more palliative surgeries (eg, a Blalock-Taussig-Thomas shunt or bidirectional Glenn/Kawashima procedure) to prepare them for definitive correction.
Case Report: We present the case of a term female neonate who was transferred to our pediatric cardiovascular intensive care unit for management of suspected congenital cardiac disease. Echocardiography confirmed the diagnosis of heterotaxy syndrome with left atrial isomerism, an interrupted inferior vena cava with azygos continuation, and a hypoplastic left ventricle with single-ventricle physiology. At 11 months of age, she underwent a Kawashima procedure with subtotal pulmonary artery ligation. She tolerated the procedure well and is anticipated to remain stable for the near future, possibly without the need for further cardiac surgery.
Conclusion: Patients with heterotaxy syndrome have congenital malformations in several organ systems, requiring lifelong coordination of care among health providers across multiple disciplines.
INTRODUCTION
Heterotaxy syndrome is a condition of abnormal lateralization of organs across the body’s left-right axis.1,2 Establishment of asymmetric tissue differentiation is a unique feature of vertebrate embryogenesis and occurs early in embryonic development.2,3 When asymmetric tissue differentiation fails, organ tissue usually specific to one-half of the body can be duplicated, while tissue from the other half can fail to develop. These developmental anomalies can result in mirror-image symmetry of normally asymmetric organs and is sometimes referred to as isomerism.2,4 Patients with heterotaxy syndrome have congenital malformations of multiple organ systems, most often the cardiovascular, gastrointestinal, lymphatic, and pulmonary systems.
Heterotaxy syndrome occurs in approximately 1 in 10,000 live births and is present in approximately 3% of congenital heart disease cases.5 Multiple genes believed to play a role in establishment of the left-right axis during embryonic development have been identified as possible contributors to the pathogenesis: ZIC3, NODAL, CFC1, ACVR2B, LEFTY2, CITED2, and GDF1.3 Growth factors responsible for establishing asymmetric left-sided tissue differentiation in mouse embryos have been shown to be interrupted in high-glucose media.6 Maternal diabetes mellitus has been identified as a risk factor for heterotaxy, namely because maternal hyperglycemia is believed to affect left-right lateralization.7
The anatomic manifestations of heterotaxy syndrome generally follow one of two patterns, referred to as right atrial isomerism (or isomerism of the right atrial appendages; these patients have two similar right atria and appendages) and left atrial isomerism (these patients have two similar left atria and appendages).
Cardiac malformations of right atrial isomerism generally show duplication of right-sided features of the heart and great vessels with an absence of left-sided features.4 Bilateral anatomic right atria are present and are the hallmark of this disease.4 Both atrial appendages are triangular, with pectinate muscle encircling the common atrioventricular junction on both sides, and a prominent terminal ridge is often present bilaterally.5 The superior vena cava can be single or bilateral.4 Transposition of the great arteries with either pulmonary stenosis or atresia is commonly seen.4 The atrial septum will usually be affected, with a primum-type atrial septal defect, secundum-type atrial septal defect, or a completely absent septum.1,2,4 The coronary sinus is often absent.4,5 Two competing sinoatrial nodes are frequently seen.2,4 Because of these atrial anomalies, pulmonary venous drainage is likely to be anomalous and obstructed.1,4 A complete atrioventricular septal defect is usually present, and a double outlet right ventricle is also a common feature.1,2,4 The inferior vena cava is usually in normal position with normal hepatic venous drainage.4
Because the spleen is a left-sided organ, patients with right atrial isomerism are more likely to be asplenic (Ivemark syndrome), and right atrial isomerism is sometimes referred to simply as asplenia syndrome.4 Other extracardiac findings in right atrial isomerism include bilateral 3-lobed lungs.4 Overall, cardiac abnormalities are much more severe in right atrial isomerism than in left atrial isomerism.4 Because of the mixing of systemic and pulmonary venous drainage, anomalous pulmonary venous drainage, and reduced pulmonary blood flow as a result of stenosis or atresia, patients frequently present at birth with cyanosis.4 Murmurs from pulmonary stenosis or ventricular septal defect may be audible, and Howell-Jolly or Heinz bodies may be present on the peripheral smear because of asplenia.4 If they have ductal-dependent cardiac lesions, these patients are often initially managed with prostaglandin E1 to keep the ductus arteriosus open until surgical intervention can be performed.
The defining cardiac finding in left atrial isomerism is bilateral left atria. Both atrial appendages will be more tubular compared to the more triangular appendages seen in right atrial isomerism, with pectinate muscle contained within the appendages.5 Venous malformations of left atrial isomerism include bilateral superior vena cavae; however, in cases of a single superior vena cava, it may be located on either the right or the left.4 Transposition of the great arteries, pulmonary stenosis, and pulmonary atresia are sometimes seen but less often than in cases of right atrial isomerism.2,4 As in right atrial isomerism, a secundum- or primum-type atrial septal defect or a completely absent septum may be present in left atrial isomerism.4 In addition, the coronary sinus and sinoatrial node can be absent, and pulmonary veins may either have normal drainage (in about half of cases) or ipsilateral-type drainage (the right-sided veins drain to the right-sided left atrium, and the left-sided veins drain to the left-sided left atrium).2,4 Approximately half of patients with left atrial isomerism have normal atrioventricular valves, with the other half having a common atrioventricular septal defect.2 Although the majority of patients have two functional ventricles, more than half have a ventricular septal defect, and some may have a double outlet right ventricle.2 The majority of patients have an interrupted inferior vena cava—usually with an absent suprarenal segment—with azygos or hemiazygos continuation to the superior vena cava.2,4,5 This last feature has the most significant differentiating power in determining left atrial isomerism, as the inferior vena cava in right atrial isomerism is almost always normal.4 Finally, bilateral common hepatic veins usually drain either to the right or left atrium.4
Extracardiac features of left atrial isomerism include multiple small spleens, usually positioned along the greater curvature of the stomach, and left atrial isomerism is sometimes referred to simply as polysplenia syndrome.2,4 Patients may have bilateral bilobed lungs, and the gallbladder may be absent.4 Patients with left atrial isomerism are less likely to be cyanotic at birth than patients with right atrial isomerism, but pulmonary overcirculation is more likely to be a complication over time.4 A murmur from a ventricular septal defect may be audible, and superior P axis on electrocardiogram may be seen due to ectopic atrial rhythm.4
Clinical findings common to both right atrial isomerism and left atrial isomerism include a midline liver, dextrocardia, symmetric mainstem bronchi, and intestinal malrotation.4 Although splenic tissue is present in left atrial isomerism, it is often nonfunctional, so patients with both right atrial isomerism and left atrial isomerism are considered to be functionally asplenic.2,4 Other gastrointestinal complications seen in heterotaxy include esophageal hiatal hernia, diaphragmatic hernia, gastric volvulus, microgastria, preduodenal portal vein, absent portal vein, biliary atresia, and pancreatic anomalies such as annular pancreas.5
We present the case of a term neonate who had heterotaxy syndrome with left atrial isomerism, an interrupted inferior vena cava with azygos continuation, and mild pulmonary stenosis who was successfully palliated with a Kawashima procedure with subtotal pulmonary artery ligation at 11 months of age.
CASE REPORT
A term female neonate born to a 34-year-old primigravida at 37 weeks’ gestation presented as a transfer from a local hospital to the pediatric cardiovascular intensive care unit at our institution. Her mother’s pregnancy was complicated by difficult-to-control diabetes and multiple fetal echocardiograms suggesting an unbalanced atrioventricular septal defect. The patient was delivered via caesarean section because of breech presentation, and the delivery was complicated by difficult extraction, with the infant requiring bag-valve-mask ventilation. She was cyanotic at delivery, and Apgar scores were 5 and 9. She was started on a prostaglandin infusion prior to transfer. Family history was negative for congenital heart disease, heterotaxy syndrome, or other genetic disorders. Prenatal genetic screening had revealed a low risk for aneuploidies; all other prenatal screens were negative.
On presentation to the cardiovascular intensive care unit, the patient’s oxygen saturation was 85%, but vital signs were otherwise normal. She was vigorous with a strong cry; her weight, length, and head circumference were appropriate for gestational age. Physical examination was notable for somewhat dysmorphic features (flat nasal bridge, flat midface, short palpebral fissures, low-set ears, cervical webbing, redundant neck folds, and widely spaced nipples), point of maximal impulse found on the right side of the chest, a normal S1 and single S2 heart sound, a 1/6 systolic ejection murmur, a liver palpable in the midline, and overlapping second digits on both feet. Electrocardiogram conducted on arrival indicated a left atrial rhythm and biventricular hypertrophy, with a repolarization abnormality. Initial arterial blood gas showed respiratory acidosis.
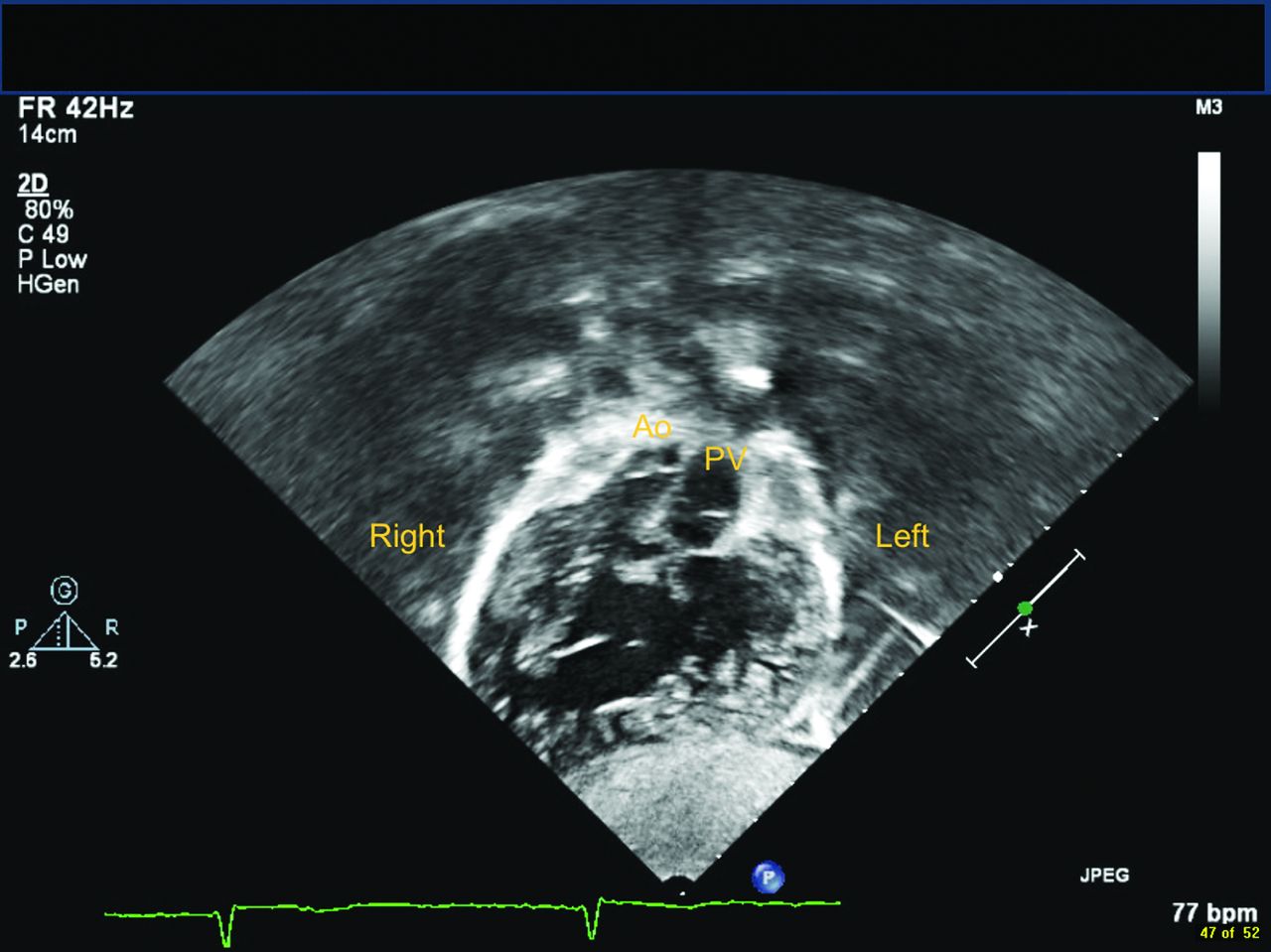
Investigations obtained at that time included a chest radiograph that revealed dextrocardia, a head ultrasound that showed a possible lesion in the basal ganglia, and an abdominal ultrasound that indicated likely polysplenia and an aorta to the right of midline. Serial echocardiograms also revealed dextrocardia (Figures 1 and 2), as well as left atrial isomerism, 4 pulmonary veins entering the right-sided morphologic left atrium, an interrupted inferior vena cava with azygos continuation, a large primum atrial septal defect, a large secundum atrial septal defect, a right ventricle–dominant unbalanced atrioventricular septal defect (Figure 2), mild common atrioventricular valve insufficiency, a hypoplastic left ventricle, a double outlet right ventricle (Figure 2), side-by-side great arteries (Figure 1), a right aortic arch with possible coarctation, mild pulmonary stenosis (Figure 1), a large bidirectional patent ductus arteriosus, and bilateral superior vena cavae with no bridging vein. Finally, an upper gastrointestinal study with small bowel follow-through showed malrotation.

Genetic testing was undertaken because of concern for aneuploidies, specifically trisomy 21 (given dysmorphic facies and atrioventricular septal defect) and Turner syndrome (given cervical webbing, wide-spaced nipples, and possible coarctation). Fluorescence in situ hybridization and single-nucleotide polymorphism hybridization were negative for aneuploidies. The patient’s head ultrasound findings raised concern for intracranial pathology related to heterotaxy or a possible intrauterine infection. Testing for congenital cytomegalovirus and toxoplasmosis was negative. Follow-up magnetic resonance imaging showed no intracranial pathology.
During the first week of her hospital stay, the patient was weaned from the prostaglandin infusion and monitored for coarctation of the aorta with patent ductus arteriosus closure. Subsequent echocardiograms revealed a closed patent ductus arteriosus without coarctation. Given the patient’s presumed functional asplenia, amoxicillin prophylaxis of 50 mg twice daily was started. The speech pathology consultant determined that the patient was at risk of aspiration because of an uncoordinated suck and recommended surgical gastrostomy tube placement. At 3 weeks of age, the patient underwent a Ladd procedure with an appendectomy to correct her malrotation and gastrostomy placement. She tolerated the procedure well. The patient’s circulation was well balanced because of her pulmonary stenosis, and no cardiac surgical interventions were needed in the subsequent month, so she was discharged home.
At 11 months of age, the patient was admitted for a Kawashima procedure with subtotal pulmonary artery ligation. She tolerated the surgery well and was weaned from mechanical ventilation the following day. During her cardiovascular intensive care unit stay, she required a brief period of noninvasive positive-pressure ventilation because of increased work of breathing but was weaned again successfully to room air. She stepped down from the cardiovascular intensive care unit on postoperative day 4 and was discharged home on postoperative day 7.
At the time of this writing, the patient was 21 months of age and had continued to gain weight. She was able to cruise but unable to walk independently, and she continued to show overlapping and curling of her toes bilaterally. She took aspirin 40.5 mg daily and amoxicillin 20 mg/kg daily for prophylaxis. From a cardiac standpoint, the majority of her systemic venous return (both superior vena cavae and inferior vena cava) was redirected to her pulmonary arteries, although her hepatic venous return drained to her right atrium. She was evaluated at 1 month, 3 months, and 6 months postoperatively by pediatric cardiology and found to be stable with appropriate growth and oxygen saturations. She is believed to be well palliated for the next several years and may not require further palliation.
Following negative genetic testing for aneuploidies, her genome underwent XomeDxSlice testing targeted to ZIC3, NODAL, and ACVR2B genes that are implicated in heterotaxy, as well as CCDC114 to screen for ciliary dysmotility. Results from these studies were normal; whole exome sequencing to identify candidate genes responsible for her phenotype may be considered in the future. Given her delay in walking and persistent toe abnormalities, she was seen by orthopedic surgery at 17 months of age and found to have sacral agenesis. She will follow up at 23 months to evaluate for possible spinal surgery. At the time of this writing, she still had her gastrostomy tube but had begun supplementing tube feeds with ad lib feeds by mouth with good results. She is followed as an outpatient by nutrition and pediatric gastroenterology. Given her cardiac history, speech and feeding difficulties, and sacral agenesis, she was referred for further neurodevelopmental workup.
DISCUSSION
Cardiac surgical intervention for patients with heterotaxy syndrome depends on ventricular physiology and circulatory balance. Because many patients with left atrial isomerism have two working ventricles, definitive biventricular repair is sometimes possible.4 For patients with single-ventricle physiology, a Fontan operation, which directs systemic venous return to the pulmonary arteries, bypassing the right heart, is considered the definitive intervention.4 Before a Fontan procedure can be completed, however, the patient usually requires a sequence of palliative surgeries to prepare for the Fontan procedure.4 These surgeries ensure that the patient has optimal ventricular function, no excessive volume or outlet obstruction causing ventricular hypertrophy, and low pulmonary resistance.4
When a patient’s cardiac lesions prevent adequate pulmonary circulation, a systemic-to-pulmonary artery shunt is often the first palliative step. One option is a Blalock-Taussig-Thomas shunt in which the right subclavian artery is connected to the right pulmonary artery.4 Alternatively, if pulmonary blood flow is too great, a pulmonary artery band can be placed to maintain circulatory balance.4 As mentioned previously, left atrial isomerism is usually associated with pulmonary overcirculation rather than inadequate pulmonary circulation. Because a Blalock-Taussig-Thomas shunt is meant to redirect more blood flow to the lungs, this intervention was not appropriate for our patient with left atrial isomerism. Additionally, our patient had mild pulmonary stenosis that helped to prevent pulmonary overcirculation, and she did not require pulmonary artery banding. Together, these features resulted in a relatively balanced circulation, allowing our patient to proceed directly to palliation with a Kawashima procedure.
Usually 2-3 months after a Blalock-Taussig-Thomas shunt or pulmonary artery band placement, once pulmonary vascular resistance has dropped, a bidirectional Glenn shunt—an end-to-side anastomosis made between the superior vena cava and right pulmonary artery—can be used to direct partial systemic venous drainage directly to the pulmonary arteries.4 For patients with left atrial isomerism who have an interrupted inferior vena cava with azygos continuation and therefore a greater portion of the systemic venous return directed through the superior vena cava, a Kawashima procedure can be performed.8 This procedure, first described by Kawashima in 1984,9 creates an end-to-side anastomosis between each superior vena cava and the ipsilateral pulmonary artery and ligates or partially ligates the main pulmonary artery; the procedure is essentially the same surgery as a Glenn shunt.8 Any previous systemic-to-pulmonary shunts are taken down at the same time.4 If circulation is relatively balanced prior to this step, as was the case with our patient, one of these procedures may be the first palliative step, meaning patients may be older at the time of their first intervention than those who require a Blalock-Taussig-Thomas shunt or pulmonary artery banding to balance their circulation.10 Oxygen saturation is carefully monitored at follow-up, as formation of venous collaterals or development of arteriovenous malformations thought to be attributable to impaired delivery of vasoconstrictive cytokines from the liver are common at this stage. Patients who undergo a Kawashima procedure usually have a follow-up procedure in which the hepatocardiac venous blood is diverted to the pulmonary circulation to prevent pulmonary arteriovenous malformations.8
When to perform a Glenn or Kawashima procedure is currently an area of some debate. The procedure has been conducted safely in patients less than 1 year old and as young as 5 months old.11 Data show that mortality rates following completion of a Glenn/Kawashima procedure are lower compared to preprocedure mortality rates; therefore, performing the Glenn/Kawashima procedure as early as tolerated can be advantageous.10,12 On the other hand, a concern is that redirection of the majority of the systemic circulation to the pulmonary vascular bed and a significant change in ventricular preload will be less easily tolerated in infants.8,11 Another concern is that younger age at the time of the Glenn/Kawashima procedure appears to be related to greater risk of developing pulmonary arteriovenous malformations, although at least one study shows low rates of pulmonary arteriovenous malformations when Glenn/Kawashima procedures are performed in patients as young as 8 months.11 Others have argued that delaying the Glenn/Kawashima procedure preserves antegrade pulmonary blood flow for as long as possible if the pulmonary artery is to be ligated later.8,13
Our patient also had subtotal ligation of the pulmonary artery at the time of her Glenn procedure, although a subject of debate is the risk and benefit of preserving antegrade pulmonary blood flow.8,13 Those in support of preserving antegrade flow cite the decreased likelihood of developing pulmonary arteriovenous malformations, improvement in oxygen saturations, a less likely need for a completion procedure to divert hepatocardiac blood to the pulmonary circulation, and the possibility for better pulmonary artery growth.11,14 Risks associated with preservation of antegrade flow include increased volume load on the single ventricle (possibly leading to valvular regurgitation), superior vena cava syndrome, arrhythmias, and a higher risk of pleural effusions.11,14
Usually 2-3 years after the second stage, the Fontan procedure may be performed, redirecting the remainder of the systemic venous return to the pulmonary arteries.4 The technique used depends on the previous palliations. In a patient who was previously palliated with a Glenn procedure, the inferior vena cava is brought to join the previous junction made with the superior vena cava and right pulmonary artery.4
Because malrotation can lead to volvulus, an upper gastrointestinal study with small bowel follow-through to the cecum is considered the gold standard for screening in all patients with heterotaxy syndrome.5 Traditionally, a prophylactic Ladd procedure is also done. The practice of performing a prophylactic Ladd procedure on patients with heterotaxy syndrome and intestinal malrotation has also become an area of debate.5 Because patients with heterotaxy syndrome experience greater postoperative morbidity and mortality than other patients with malrotation, some consider the complications associated with the Ladd procedure to be a greater risk than development of volvulus and have suggested a watchful waiting policy for asymptomatic patients.15,16
Because patients with heterotaxy syndrome are functionally asplenic, they are managed with vaccination against Haemophilus and pneumococcal species, as well as antibiotic prophylaxis (usually with penicillin, amoxicillin, or—in the case of penicillin allergy—erythromycin).5 However, incidence of sepsis is still high in patients with heterotaxy syndrome, with cases showing nosocomially acquired organisms more prevalent (eg, Klebsiella, viridans streptococci, Staphylococcus sp, and Enterococcus sp); meanwhile, sepsis caused by more traditionally implicated pathogens has greatly decreased.17 As our patient with left atrial isomerism is considered to be functionally asplenic despite the presence of splenic tissue, she was vaccinated against Haemophilus influenzae type B and received the pneumococcal 13-valent conjugate vaccine at 2, 4, 6, and 15 months. She also continues to receive amoxicillin 20 mg/kg daily.
Heterotaxy syndrome has been linked to ciliary dysfunction.3,5 This link has been postulated to contribute to the greater incidence of postsurgical respiratory complications seen in heterotaxy syndrome patients.5,18 Screening for ciliary dysfunction and then treatment with inhaled beta agonists may improve survival.19 In our patient’s case, genetic screening for CCDC114 mutations was conducted, as this gene has been shown to cause ciliary dysmotility and primary ciliary dyskinesia20; however, results were negative. The decision was made to screen via further genetic testing or ciliary biopsy if she began to experience recurrent sinopulmonary infections. To date, screening has not been necessary.
Surgical outcomes for the Blalock-Taussig-Thomas shunt and the Glenn, Kawashima, and Fontan operations have improved over time, but overall, the prognosis for patients with heterotaxy syndrome is significantly worse than for patients with other congenital heart diseases,5,21 possibly because the factors conferring increased mortality risk following these procedures—such as neonatal presentation requiring surgery,22 pulmonary venous obstruction or stenosis,10,22,23 atrioventricular valve dysfunction,21-23 unplanned reoperation,21 total anomalous pulmonary venous return,21 extracorporeal membrane oxygenation use,21 arrhythmia,10 and longer duration of intensive care unit stay10—are more likely to be seen in patients with heterotaxy syndrome. In addition, functional asplenia in these patients carries a higher risk of sepsis,10,17 and gastrointestinal issues can lead to poor feeding and poor weight gain.10 However, studies that have compared short- and long-term prognoses show that while interstage mortality related to these interventions (particularly between the first palliative cardiac procedure and the second) shows a significantly worse prognosis for patients with heterotaxy syndrome, survival rates following cavopulmonary anastomosis procedures, as well as long-term survival, are comparable between patients with heterotaxy syndrome and their peers with other congenital heart diseases.10,21,24,25
While some patients who undergo these surgeries as children report occupational limitations as adults, overall, self-reported health is good in the majority of patients.5 In a study comparing patients aged 6-18 years with heterotaxy syndrome to patients with other congenital heart diseases, patients with heterotaxy syndrome had higher rates of rhythm abnormalities on electrocardiogram and a greater number of anomalous findings on echocardiogram, but these patients showed no difference in exercise performance, brain natriuretic peptide levels, or health status questionnaire scores.25
CONCLUSION
Patients with heterotaxy syndrome have multiple congenital cardiac anomalies, usually requiring surgical correction at a young age. In addition, patients with heterotaxy syndrome may have congenital malformations in several other organ systems, making management of cardiac conditions more complex compared to patients with other congenital cardiac diseases and requiring coordination of care among health providers across multiple disciplines. Our patient’s case provides a clear illustration of the cardiac and extracardiac concerns for patients with left atrial isomerism, as well as the importance of a balanced pulmonary circulation to good therapeutic outcomes. Understanding the common anatomic variations and health challenges associated with heterotaxy syndrome is essential for providing optimal care.
This article meets the Accreditation Council for Graduate Medical Education and the American Board of Medical Specialties Maintenance of Certification competencies for Patient Care and Medical Knowledge.
ACKNOWLEDGMENTS
The authors have no financial or proprietary interest in the subject matter of this article.
- © Academic Division of Ochsner Clinic Foundation
REFERENCES
In this issue









{kind=link}
{kind=link}
Jump to section
Cited By...
- No citing articles found.