
West Nile virus (WNV), a member of the Flavivirus genus family Flaviviridae, was first isolated in 1937 from the blood of a female patient participating in a malaria study in the West Nile district of northern Uganda. Until 1999, the virus was found primarily in the eastern hemisphere with wide distribution in Africa, Asia, the Middle East, and Australia and has been responsible for significant epidemics, notably in Israel (1950s), France (1962), South Africa (1974), Algeria (1994), Romania (1996), and Italy (1998) (1). However, the outbreak of WNV illness in the New York City metropolitan area in the Summer of 1999 emerged as the first epidemic in the western hemisphere. In 1999, WNV was responsible for 62 cases of encephalitis, meningitis, or both. From 2000–2002, the virus extended its range throughout much of the eastern United States with 3389 reported cases of human WNV-associated illness from 37 states and the District of Columbia, compared with 149 during 1999–2001 (2). Five of the states (Illinois, Michigan, Ohio, Louisiana, and Texas) accounted for as much as 67% of the reported West Nile meningeoencephalitis cases. It is expected that WNV will continue to extend its range within the western hemisphere. Sequence analysis of the New York City isolates from animals and humans showed that the strains in North America have a high degree of homology with the 1998 strains from Israel, suggesting that the outbreaks in the US resulted from the introduction of a virus that has been circulating in the Middle East since at least 1998 (3).
The genus Flavivirus comprises about 70 viruses classified on the basis of their serologic and genomic relatedness. WNV is a member of the Japanese encephalitis (JE) serocomplex. Other members of this complex include St. Louis encephalitis in the western hemisphere, Murray Valley encephalitis in Australia, and Rocio virus in South America (Table). Phylogenetic analysis of WNV strains has identified two genetic lineages: I and II (4). All the strains associated, thus far, with outbreaks of human and equine disease belong to lineage I and have a wide distribution including Africa, Asia, Europe, and North America. Lineage II strains of WNV are restricted to endemic enzootic infections in Africa and have not been associated with human encephalitis cases.
Mosquito-borne flaviviruses causing human disease
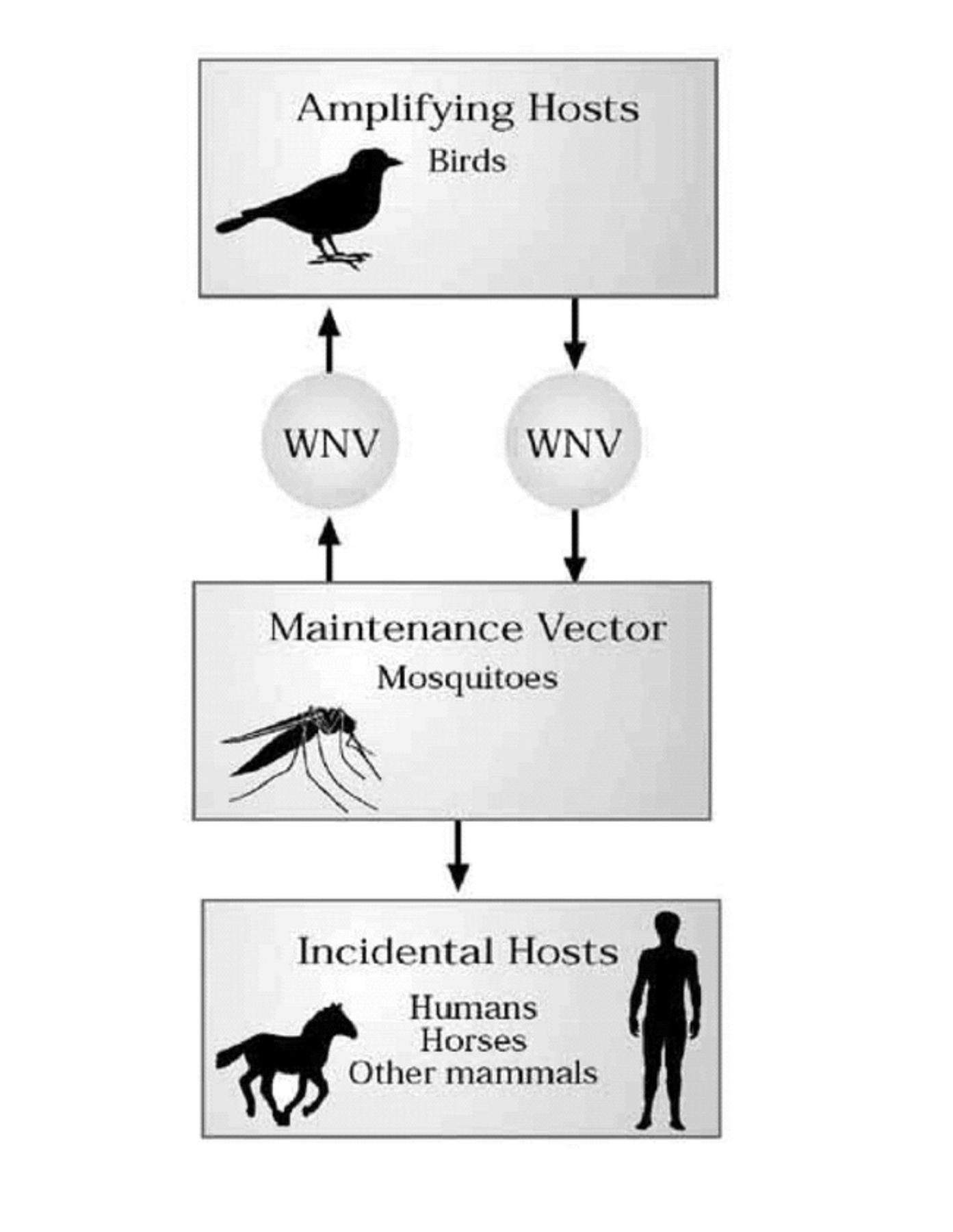
TRANSMISSION CYCLE OF WNV
WNV is maintained in nature in a mosquito-bird cycle primarily involving Culex species of mosquitoes. The three species that were among the most important WNV vectors during the 2002 epidemic in the United States were Cx. pipens, Cx. quinque-fasciatus, and Cx. restuans (2). However, the virus was isolated from 33 mosquito species during the 1999–2002 epidemics. For the mosquito to transmit the virus by bite, the virus must replicate throughout the body of the mosquito and the salivary gland after it feeds on a viremic host. Viral amplification occurs in the bird-mosquito-bird cycle (Figure 1). Infected birds commonly survive their infection and develop immunity, although individuals of some species become ill and die. WNV has been shown to infect more than 100 bird species in North America alone (5). However, the virus is particularly virulent in species belonging to the family of Corvidae (e.g. crows and blue jays). Particularly high mortality rates are noted among infected crows and blue jays (∼90% of the infected dead birds) whereas the remaining ∼10% belonged to 92 other species in the 2002 outbreak of WNV infection in the US.

Transmission cycle of West Nile virus.
The first indicators of WNV infection are usually dead birds in an area before the recognition of human illnesses. Sixty two percent of the counties in 44 states and the District of Columbia reported infected dead birds during WNV activity in 2002, compared with only 4% of the counties reporting a case of human infection. Dead-bird-based surveillance programs are also very important tools for monitoring the geographical spread of WNV. During the 1999–2002 epidemics, the virus exhibited rapid geographical expansion from 4 states in 1999 to 44 states and the District of Columbia in 2002.
A broad range of mammalian species are susceptible to natural infection with WNV following infectious mosquito bite(s). In the US during 1999–2001, nine mammalian species (humans, horses, cats, rabbits, skunks, squirrels, chipmunks, and two species of bats) were found to be infected with WNV (6). However, naturally acquired disease has been conclusively demonstrated in human beings and equines. Humans and domestic animals are considered incidental or dead-end hosts because they apparently do not develop viremia high enough to infect mosquitoes and contribute to the transmission cycle (Figure 1).
GENOME STRUCTURE
The WNV is a spherical particle ∼50 nm in diameter and consists of a host-derived lipid bilayer membrane surrounding a nucleocapsid core. The nucleocapsid contains a single-stranded positive-sense RNA genome 11,029 nucleotides in length and consisting of a 92-nucleotide long 5′ non-coding region followed by a single open reading frame of 10,301 nucleotides coding for 3 viral structural proteins and 7 non-structural proteins, and a 631-nucleotide long 3′ non-coding region (Figure 2) (3, 7). The non-translated nucleotides regions of the flaviviral genomes appear to be important for regulating viral translation, replication, and packaging. The genome is translated into a large polyprotein which is processed to individual proteins by host cell and viral proteases. The viral serine protease, NS2B-NS3, and several cell proteases then cleave the polyprotein at multiple sites to produce 10 mature viral proteins. NS5 located at the C terminus of the viral polyprotein possesses RNA-dependent RNA polymerase activity. The N-terminal region of NS5 also contains a methyltransferase domain that is thought to function in the methylation of the type 1 cap at the 5′ terminus of the viral genome. The majority of the nonstructural proteins is multifunctional and mainly involved in the formation of active RNA replication complexes.

WNV RNA genome consisting of 5′ non-coding region (NCR), a single open reading frame coding for three structural proteins and seven non-structural proteins, and a 3′ NCR.

The viral structural proteins (capsid, membrane [prM/M], and envelope) are encoded within the amino-terminal one-third of the polyprotein. The capsid protein (C) packages the RNA genome. The viral envelope is composed of the envelope (E) and membrane (M) proteins. Virions may also contain variable amounts of prM (the precursor of M) glycoprotein. However, virions with preM-E complexes are less infectious than mature virions (8). The E glycoprotein is essential for virion assembly and mediates binding to cellular receptors. It is also the primary target of virus-neutralizing antibodies.
REPLICATION CYCLE
Flaviviruses replicate in a variety of insect, mammal, and avian cells. After binding to specific, but as yet unknown cell receptors, virions enter cells via receptor-mediated endocytosis followed by fusion between the viral and cell vesicle membranes, releasing the nucleocapsid into the cytoplasm where it is translated into a single polyprotein (7). The polyprotein is cleaved at multiple sites by viral and cellular proteases to produce the 10 mature viral proteins. The viral RNA-dependent RNA polymerase, NS5, with other viral nonstructural proteins and possibly cell proteins, copies complementary minus strand RNAs from the genomic RNA. The minus strand RNAs, in turn, function as templates for the synthesis of new genomic RNAs. The newly synthesized RNAs have multiple functions during the viral replication cycle. The RNAs could serve as templates for the synthesis of minus strand RNAs, mRNAs for synthesis of the viral polyprotein, and as substrates for virion assembly. Progeny virions are transported to the cell surface and are released by exocytosis. In WNV-infected mammalian cells cultures, the release of progeny virions is normally seen 10–12 hours after infection.
VIRUS-HOST INTERACTIONS
Several factors influence the outcome of WNV infection in the human and animal hosts including virus strain, age, immune status, and genetic susceptibility. Advanced age is a major risk factor for WNV disease and death, especially in patients older than 70 years of age. Among the 13% fatal cases in the outbreaks in US during 1999–2000, median age of patients was 75 years (range 44–90 years), and 89% were older than 60 years (6). Among the non-fatal cases, median age of patients was 65 years (range 5–90), and 59% were older than 60 years.
In mice, WNV is neurotropic and produces experimental lesions which are similar to those reported in natural infections of humans and horses. However, variation exists in neurovirulence and neuroinvasion among different viral strains. A comparison of two WNV strains, one that caused an outbreak in 1998 in Israel (similar to the strain isolated in New York) and another that was isolated from Aedes aegypti mosquitoes in Senegal in 1990, showed that the Israeli isolate was much more lethal to mice at a much lower dose than the Senegalese strain, suggesting that epidemic WNV was more pathogenic than the enzootic virus (9).
In addition to strain differences, passage history itself could affect the extent of neuroinvasion. For example, prolonged passage of the Israeli isolate in mosquito cells resulted in loss of neuroinvasiveness (10). Although these viruses had a mutation in their E protein region that produced an N-linked glycosylation site, this mutation did not directly correlate with loss of neuroinvasiveness. Experimental evidence suggests that additional mutations in E as well as mutations within other regions of the viral genome also affect neuroinvasiveness and are required for attenutation (10).
Innate resistance to flavivirus-induced disease was demonstrated in mice in the 1920s. Subsequent breeding studies have shown that the alleles of a single Mendelian dominant gene, Flv, can determine whether WNV infection is lethal (7, 11). The majority of the inbred mouse strains currently in use are susceptible to WNV disease, whereas most wild mice are resistant. The inbred strains carrying the Flv resistance allele found in resistant strains maintain resistance to WNV-induced disease. However, disease-resistant mice are not resistant to WNV infection. These mice produce lower virus titers and the spread of infection is slower, which gives the host defense system sufficient time to effectively eliminate the infection. The Flv gene is also known to confer resistance to other mosquito-borne flaviviruses, including yellow fever and dengue viruses, and to certain tick-borne viruses.
IMMUNE RESPONSES TO WNV
Significant differences exist in the prevalence of background immunity in human populations. In some endemic areas of Africa it ranges from roughly 50% in children to almost 90% in adults. By contrast, recent epidemics in North America and Europe did not result in significant levels of background immunity in post-epidemic areas. Studies on WNV reactive IgM persistence have shown that IgM may remain detectable for several months in some WNV encephalitis survivors (12). In a recent study, the percentage of WNV encephalitis patients with detectable serum IgM antibodies at 6 months was 65% and dropped to 15% (<65yrs: 0%; >65yrs: 15%) at 12 months (13). The duration of IgM antibody in cerebrospinal fluid (CSF) is not known. However, a positive serum test for IgM antibody to WNV might suggest IgM persistence in CSF, or signify a persistent or relapsing CNS infection. Persistent or relapsing infection has been seen in humans and nonhuman primates infected with WNV and other members of the Japanese encephalitis complex (4, 14).
The E protein is the prime target of virus-neutralizing antibody. In particular, structural domain III of E has been postulated to primarily contain the receptor-binding region. Antibodies binding to domain III block virus attachment to cells. Patients with WNV infection produce antibodies that recognize the recombinant E protein (15). In one study, immunization with recombinant E protein protected mice against experimental WNV infection (15). Moreover, a single intramuscular injection of DNA encoding the WNV virus prM and E proteins induced protective immunity against WNV infection in mice and horses (16). Antibodies to several other viral proteins are also detected in flavivirus-infected hosts. Dominant T- and B-cell epitopes have been mapped to NS1 and NS3 proteins of several flaviviruses, although immune responses to C protein and other nonstructural proteins are also seen (11). A DNA vaccine encoding the WNV C protein induces strong antigen-specific humoral and cellular immune responses in DNA vaccine-immunized mice (17). Other immunization strategies that have been evaluated include a killed virus veterinary vaccine, an attenuated live virus chimera consisting of yellow fever 17 D vaccine with the prM and E genes replaced with those of WNV, and passive immunization with WNV-immune serum (18–20). Each of the products protected the animals from illness and death in a hamster model of the WNV disease (20). In another study, an attenuated WNV variant, WNI-25A, has been shown to induce protective immunity in geese against a WNV isolate that closely resembled the virus isolated during the 1999 New York epidemic (19). WNI-25A is a variant that lost the neuroinvasion trait after serial passages in mosquito cells. Protection from WNV challenge has been shown in experimental hosts infected or immunized with closely related flaviviruses. Immunization of bonnet macaques with Japanese encephalitis virus protected the animals against clinical signs of WNV disease (21). A study on susceptibility of domestic pigs to WNV showed that Japanese encephalitis virus infection boosted the already existing WNV antibodies (22).
CONCLUSION
WNV has a widespread distribution in Africa, West Asia, the Middle East, and Europe. However, the introduction of WNV into the US for the first time in 1999 demonstrated the ease with which new and emerging pathogens can jump continents and hemispheres at an increasing rate. From 1999–2002, WNV rapidly spread throughout much of the eastern part of the US and will likely continue to spread into the western parts over the next several years, primarily via the movement of viremic birds. Thus, detailed epidemiological, ecological, and virological studies are necessary to address the many questions that the recent spread of WNV in the US has raised, such as 1) the identification of local mosquito species in the transmission of WNV to birds, humans, equines, and possibly to other animals; 2) the importance of non-mosquito transmission, such as direct bird-to-bird transmission or through predators that feed on infected birds; 3) the contribution of infected migratory bird species that survive infection; 4) the role of mosquitoes and birds in the over-wintering of WNV; and 5) the influence of local environmental conditions on the magnitude of WNV transmission.
WNV, like several other flaviviruses, cycles between insect vectors and vertebrate hosts in nature. Any cellular proteins used by the virus are expected to be highly conserved among divergent species. Identification of these proteins and analysis of their functions in virus attachment, entry, replication, virulence, and disease potential is critical to our understanding of the virus-host interactions at molecular levels and for the development of novel antiviral therapies.
No vaccines against WNV are currently available for human use, although several laboratories are conducting research. Epidemiological and ecological studies over the next few years will determine the overall risk of acquiring WNV virus infection and subsequent illness, and whether a vaccine-based approach towards disease prevention is justified. However, effective surveillance systems and vector mosquito control programs will continue to remain central to the prevention of WNV epidemics.
- Ochsner Clinic and Alton Ochsner Medical Foundation
REFERENCES
In this issue








{kind=link}
{kind=link}
{kind=link}
Jump to section
Cited By...
- No citing articles found.