
INTRODUCTION
Much of our current understanding of oncogenesis arose from a 1911 finding by American pathologist Peyton Rous that cell-free extracts of a chicken fibrosarcoma transmitted the disease to other chickens, indicating that the tumor was due to a “transmissible agent.” More than 40 years later, this led to the discovery of Rous sarcoma virus (RSV), named for its discoverer (1). In 1966, at the age of 87, Rous was awarded a Nobel Prize in recognition of his discovery of tumor-inducing viruses. The evidence that RSV is a retrovirus (RNA) tumor came from the Nobel Prize-winning discovery of reverse transcriptase by Howard Temin (2) and David Baltimore (3), which indicates that the viral genome is reverse transcribed into a DNA copy that becomes integrated into the host chromosome DNA as a provirus. This new genetic information acquired by the host serves as a source of virus production.
DISCOVERY OF CANCER GENES
The malignant changes induced by RSV are the consequence of the expression of a single viral oncogene, termed v-src for viral sarcoma, v-src is a 60 kDa nonreceptor tyrosine kinase that has the unusual specificity of phosphorylating tyrosine residues of critical cellular substrates, resulting in the activation of oncogenic transduction pathways. Expression of v-src in susceptible avian or mammalian cells leads to transformation in vitro and tumorigenesis in vivo (4,5). The discovery that RSV contained a defined gene for transformation opened a new frontier in the search for genes that might be involved in cancer. Eventually, the pioneering work of Mike Bishop and Harold Varmus led to the discovery of the normal cellular counterpart of the oncogene v-src, termed c-src, which in 1989 earned them the Nobel Prize in Physiology and Medicine. The term “protooncogene” was coined to describe the new types of genes, distinguishing them from the oncogenes.
STRUCTURE AND REGULATION OF SRC KINASES
The discovery that c-src is the normal cellular counterpart of the v-src led to the efforts to unveil the structural and functional differences between the two. Like its viral counterpart v-src, c-src also encodes a non-receptor tyrosine kinase. However, unlike v-src, the tyrosine kinase activity in c-src is tightly regulated and, for the most part, is maintained in an inactive state, activated transiently in response to specific stimuli. The main difference is found in the structure of their C-terminal regions, c-src has seven domains: an N-terminal membrane-association domain (SH4); a unique domain; SH3 and SH2 domains; an SH2-kinase linker domain; a tyrosine kinase domain (SH1); and a short c-terminal domain that plays a significant role in regulation of Src kinase activity (Fig. 1). The catalytic activity of Src family kinases is determined by the phosphorylation status of key tyrosine residues in their molecules. Positive regulation of human c-src protein occurs through autophosphorylation of tyrosine (Tyr) 419 (Tyr 416 in chickens), which is located in the SH1 domain and is required for optimal activity. Following autophosphorylation, the enzyme is stabilized in its active form. The negative regulation involves binding of phosphorylated Tyr 530 (Tyr 527 in chickens) in the human c-src protein to its own SH2 domains, which results in inactive conformation. Src with phosphorylated Tyr 530/527 cannot undergo Tyr 419/416 autophosphorylation. Under basal conditions in vivo, 90%–95% of Src is phosphorylated at Tyr 530/527. Dephosphorylation results in the unfolding of Src family kinases (SFKs), exposing Tyr 419/416, which requires phosphorylation for full activity. The loss of Tyr 530/527 or its dephosphorylation leads to stimulation of Src catalytic activity.
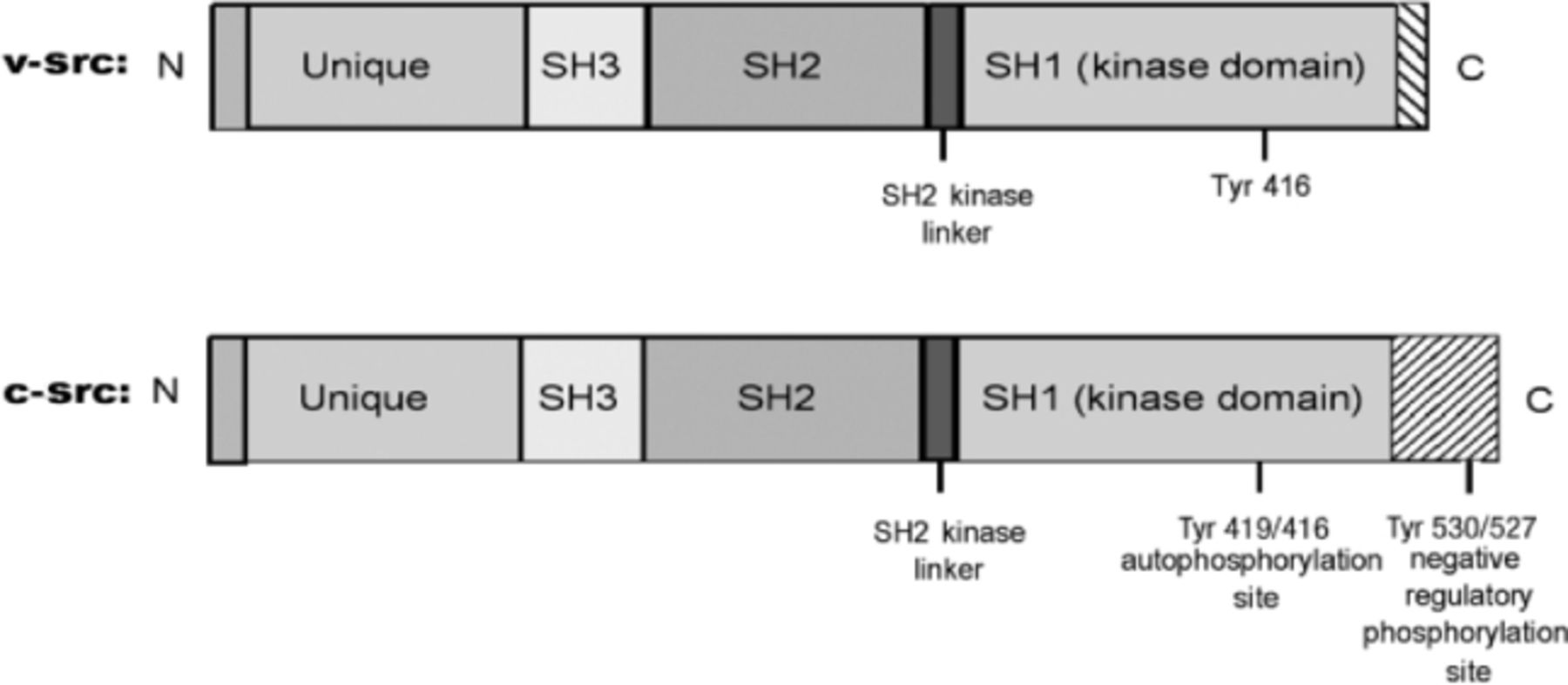
Domain structures of v-src and c-src proteins. Note the difference in their C-terminal regions. The first number is for human and the second for chicken c-src proteins.
Although the structure of v-src and c-src is similar, the two proteins differ, primarily in their regulatory C-terminal regions. V-src lacks the 19 amino acids present in the regulatory C-terminal region of c-src. The negative regulatory phosphorylation site is thus deleted, resulting in high levels of kinase activity and transforming ability (Fig. 1). The kinase activity of v-src and its transforming ability are much higher than c-src. The first genetic evidence that activating Src mutations may have a role in advanced human colon cancers came from the observation that in metastases of human colon cancer, c-src has mutations just in this region in a small subset of patients (6). However, such mutations were not detected in other studies (7), suggesting that genetic activation of c-src is a rare occurrence. Rather, Src protein kinase levels and Src kinase activity are frequently elevated in different cancers, including colorectal, breast, pancreatic, lung, neural, ovarian, and other common human cancers (8). Several possible mechanisms have been proposed, including direct or indirect interactions with receptor tyrosine kinases or defects in the phosphorylation of the carboxyl-terminal residue (Tyr 530/527), the primary phosphorylation site involved in the inactivation of c-src, suggesting that the process is multifactorial (8,9).
To date, a total of 11 distinct members of the Src family of kinases have been identified in humans (10). These include Blk, Brk, Fgr, Frk, Fyn, Hck, Lck, Lyn, Src, Srm, and Yes. They share extensive homologies in the catalytic domains but little homology in the amino-terminal regions, which may contribute to their unique biological functions (11). Among all family members, c-src is most frequently implicated in human cancers, including those of the colon, breast, pancreas, and brain. Lyn, one of the members of this family, is the predominant SFK involved in cell survival in heamatopoietic cell lineages (12). However, recent studies indicate that the Lyn kinase may be involved in the pathogenesis of certain solid and hematologic malignancies.
LYN KINASE: ROLE IN HEMATOLOCIC MALIGNANCIES
Lyn is over-expressed and activated in freshly isolated malignant B lymphocytes from the peripheral blood of B cell chronic lymphocytic leukemia (B-CLL) patients compared with normal B lymphocytes. Addition of the Lyn inhibitors to B-CLL cell cultures induced apoptosis, suggesting that Lyn contributed to the defective apoptosis of the leukemic cells (13). Lyn is also the most predominant SFK in primary leukemic cells from acute myeloid leukemia (AML) patients (14). Mutations found in Flt3, a hematopoietic growth factor receptor, in AML cause constitutive Lyn phosphorylation and activation. Targeting Lyn in mice transplanted with Flt3 mutation-expressing murine myeloid cells blocked the onset of tumors and decreased the size of established tumors (15).
Chronic myelogenous leukemia (CML) is characterized by the Philadelphia (Ph) translocation t(9;22)(q34;q11.2), which results in the formation of a bcr-abl fusion gene that translates an oncogenic protein-tyrosine kinase with transforming activity for hematopoietic cells. Imatinib mesylate (Gleevec; Novartis, Basel, Switzerland) is a potent and selective inhibitor of Bcr/abl tyrosine kinase and is the first choice treatment for all newly diagnosed CML patients. However, nearly 4% of the newly diagnosed patients on imatinib develop resistance to the drug. Studies have shown that Lyn expression is elevated in peripheral blood lymphocytes taken from patients with advanced CML that progressed on imatinib mesylate (Gleevec) therapy. Antisense oligonucleotides or short interfering RNA (siRNA) that suppressed Lyn expression had potent inhibitory effects on the growth of imatinib-resistant human CML cells (16,17). Lyn activation also plays a functional role in upregulation of Bcl-2, an antiapoptotic protein, in CML cells displaying Bcr/Abl-independent forms of imatinib resistance (18). These results clearly supported a role for Lyn kinase in the growth and apoptotic protection of imatinib-resistant CML cells. The novel observation that overexpression of Lyn is associated with imatinib resistance prompted investigation into the use of dual Bcr-Abl/SFK inhibitors. Several of these inhibitors from various chemical classes have been found to be 100 to 300 times more effective than imatinib (19).
LYN KINASE: ROLE IN SOLID TUMORS
Lyn overexpression and activation in colon carcinoma cells confers chemoresistance (20). Lyn kinase activity is significantly elevated in glioblastoma tumor biopsies compared with non-neoplastic brain biopsies. In the tumors, Lyn kinase activity accounts for >90% of the total Src kinase activity, compared with only ∼30% in non-neoplastic brain tissues. These results suggested that Lyn kinase promoted malignant transformation of glioblastoma tumors (21).
Lyn is expressed in a majority of primary human prostate cancers (22). Inhibition of Lyn activity using a sequence-based Lyn inhibitor peptide decreases proliferation of hormone refractory prostate cancer cell lines and significantly reduces tumor growth in a tumor explant model. Thus, Lyn may serve as a prime target for the treatment of malignancies that are linked to deregulated Lyn expression/activation.
CONCLUSION
The pioneering work on the Rous sarcoma virus was directly responsible for the discovery of cellular protooncogenes, which has been fundamental in shaping our understanding of cellular growth control. Over the last 50 years, advances in the understanding of cancer biology have made it increasingly clear that the activation of oncogenes often results in the deregulation of complex cell-signaling pathways that promote proliferation, apoptotic-resistance, angiogenesis, and metastasis. Hence, the signaling components of these deregulated pathways provide selective targets to guide therapeutic decisions.
Lyn kinase, once considered an exclusive B-cell signaling molecule involved in the pathogenesis of hematologic malignancies, has more recently been shown to play a role in solid tumors. Thus, one can envision targeting a wide range of malignancies with the same inhibitor as a single agent or in combination with other therapies.
Imatinib mesylate is the first successful example of a tyrosine kinase inhibitor for CML. However, the initial striking success of this drug has been clouded by the development of clinical resistance that mostly coincides with the activation of SFKs. This prompted the development of dual Src and Abl inhibitors such as dasatinib (Bristol-Myers Squibb, New York, NY), which targets Bcr-Abl and multiple SFKs, and INNO-406 (Innovive Pharmaceuticals, New York, NY), which targets Bcr-Abl and a single SFK Lyn kinase. In preclinical studies, these inhibitors and others were several times more effective than imatinib. Although originally designed for CML, dasatinib also shows activity against epithelial tumor cells, including human prostate and breast cancer cells (23). Dasatinib has recently been approved by the US Food and Drug Administration for the treatment of imatinib-resistant and imatinib-tolerant CML, as well as the treatment of therapy-resistant Ph+ acute lymphoblastic leukemia (24). Thus, the dual Bcl-Abl/SFK inhibitors represent a major success in targeted chemotherapy and also have potential as therapeutic agents for cancers harboring activated SFKs.
The more we learn about the deregulated cell signaling pathways induced by the activation of oncogenes, the better we are able to design new strategies to improve cancer treatment. This is especially critical for the subpopulation of patients who fail to respond to conventional chemotherapies. We continue to learn from clinical experience that patients may respond much better to pathway-driven therapeutics than to standard cytotoxic chemotherapies. Thus, deciphering the critical pathways that the oncogenes or inactivation of tumor suppressor genes regulate will translate into greater benefits for cancer patients by a more precise matching of therapy with disease mechanism(s).
- Ochsner Clinic and Alton Ochsner Medical Foundation
References
In this issue








{kind=link}
Jump to section
Cited By...
- No citing articles found.